




Idiopathic pulmonary fibrosis has a median survival time after diagnosis of 2–5 years. The main goal of treating IPF is to stabilize or reduce the rate of disease progression. Nintedanib and Pirfenidone have been a breakthrough in the management of IPF. Here we evaluated the effectiveness of Pirfenidone and Nintedanib in a population of IPF patients diagnosed in the last 12 months at Florence ILD Referral Centre.
MethodsIn the last 12 months, 82 IPF patients (66 male, mean age 78.3±23.8 years) were diagnosed and started antifibrotic therapy with Pirfenidone or Nintedanib. Their clinical and functional details were analyzed retrospectively at time 0 and after 6 and 12 months of therapy.
ResultsThe median age of the patients treated with Nintedanib was higher than that of the Pirfenidone group (p<0.0001). The most common symptoms at disease onset were exertional dyspnoea and dry cough with no differences between the two groups (p<0.05). All IPF patients manifested bibasal crackles at the time of diagnosis. No significant differences in FVC, FEV1, TLC and DLCO were found at time 0 or after 6 months between patients treated with Pirfenidone and Nintedanib (p>0.05). After 1 year, lung function test parameters of patients treated with Pirfenidone had remained stable from baseline.
DiscussionThis study emphasizes that both antifibrotic drugs appeared to be a good therapeutic choice in terms of functional stabilization, also in older patients.
Idiopathic pulmonary fibrosis (IPF) is a chronic progressive interstitial lung disease (ILD) of unknown aetiology, limited to the lungs, and characterized by a usual interstitial pneumonia (UIP) histopathological pattern.1,2 IPF accounts for approximately 55% of all idiopathic interstitial pneumonias (IIP) and 25% of all interstitial lung diseases (ILD) with about 35,000 new diagnoses per year in Europe and 5 million in the world.3 It has an annual incidence of 0.22–7.4 per 100,000 persons and a prevalence of 1.25–23.4 cases per 100,000 of population and is more frequent in patients between 65 and 79 years of age.3 It is a disabling and fatal disease with a poor prognosis.1–11 Five-year survival is 20–40%, which is lower than for breast cancer, colorectal cancer and pulmonary arterial hypertension.1–5 Because there is no known cure, the goal of IPF treatment is to stabilize or reduce the rate of disease progression.6 Two therapies for IPF, Nintedanib and Pirfenidone, were recently approved by EMA respectively in …… and FDA 4 years ago. The new drugs have been a breakthrough in the management of IPF.6 The world-wide phase III clinical trials ASCEND, CAPACITY 004 and 006 demonstrated for the first time that Pirfenidone reduced IPF progression and brought long-term benefits to these patients.6–10 The ASCEND trial assessed Pirfenidone efficacy after 52 weeks, while the CAPACITY study determined the effects of the drug over an observation period of 72 weeks.8 These trials found that Pirfenidone treatment significantly reduced the decline in forced vital capacity, improving the 6-min walking test distance (6MWD) by 50m.7 In addition, progression-free survival analysis showed that the risk of IPF progression fell by 38% in the Pirfenidone group compared to the placebo-group. The risk of IPF mortality during treatment declined by 60% in the group of patients treated with Pirfenidone, compared to the placebo group, while the risk of mortality for all causes fell by 37%.10 Likewise, phase II and III clinical trials revealed that Nintedanib slowed progression of the disease, reducing the decline in FVC,12,13 while the TOMORROW study showed a 68% reduction in annual FVC decline rate for Nintedanib-treated patients compared to a placebo group and a reduction in the number of acute exacerbations in treated compared to untreated patients. The INPULSIS I and II studies consistently demonstrated the efficacy of Nintedanib in reducing disease progression with a significant slowing down in the annual decline in FVC.12,13 The Italian Medicines Agency AIFA approved Pirfenidone for use in Italy in June 2013, while Nintedanib was authorized in April 2016. Regional Referral Centres for ILD can prescribe these therapies for patients with IPF.
In the present study we evaluated the effectiveness of Pirfenidone and Nintedanib in a population of patients followed at our hospital last year. The clinical and functional characteristics of IPF patients treated with Pirfenidone or Nintedanib were recorded at the start (time 0) and after 6 and 12 months of treatment to define the effectiveness of antifibrotic therapy and compare it with data in the literature.
Population and methodsIn the last 12 months, 82 IPF patients (66 male, mean age 78.3±23.8 years) have been diagnosed with interstitial lung diseases (ILD) at AOUC Careggi Referral Centre and started antifibrotic therapy with Pirfenidone or Nintedanib. The clinical and functional details were analyzed retrospectively from demographic data, family history for pulmonary fibrosis, smoking, occupational/environmental exposure and comorbidities. The following parameters were recorded: age, sex, BAL differential cell count, BAL lymphocyte phenotype, lung function tests, oxygen desaturation during 6min walking test, and PaO2 values measured by blood gas analysis. All these parameters were entered in a database together with functional, radiological, histological and immunological data. Lung function test parameters were performed according to ATS/ERS guidelines collecting percent predicted FEV1, FVC, TLC and DLCO.14 IPF diagnosis was based on clinical radiological parameters in 70/82 IPF patients. Chest X-rays in the posterior-anterior and lateral projections and high-resolution computed tomography of the chest were done in all patients. Diagnosis was formulated in the context of a multidisciplinary meeting.
Histological diagnosis of IPF/UIP was obtained in 12 patients with HRCT signs of IPF with no clear radiological honeycombing; four patients underwent transbronchial lung cryobiopsy, a single patient underwent non-intubated thoracic surgery performed under spontaneous ventilation (AWAKE VATS) and seven patients a VATS lung biopsy.
In a subgroup of patients bronchoscopy with bronchoalveolar lavage was performed with informed consent of patients in order to exclude other interstitial lung diseases. Briefly, samples were obtained by instillation of four aliquots of 50ml saline solution by videofiberoptic bronchoscope. Each aliquot was immediately gently aspirated. The first BAL sample was kept separate from the others and was not used for immunological tests. Sub-samples were cultured for microbes, fungi and viruses to exclude infections. Cells were separated by centrifuge and the fluid fraction was frozen for enzyme assays. Differential cell counts were done. Lymphocyte phenotype was analyzed by flow cytometry using anti-CD3, CD4 and CD8 monoclonal antibodies.
The patients included in the study had not been treated with steroids or other immunosuppressants at the time of BAL. They were monitored every 3 months from onset at our regional interstitial lung disease referral centre. All gave written informed consent to participation in the study.
Patients with IPF were treated with Pirfenidone or Nintedanib according to Italian national drug inclusion/exclusion criteria. Nintedanib inclusion criteria in Italy were age ≥40 years, diagnosis of idiopathic pulmonary fibrosis according to international guidelines, FVC >50% of predicted and DLCO >30%. Exclusion criteria for Nintedanib treatment in Italy were ALT, AST >1.5× ULN, total bilirubin >1.5× ULN, high risk of bleeding, INR >2, PT, PTT >150% of ULN, major surgery scheduled in the next 3 months or high risk of thrombosis. Pirfenidone inclusion criteria in Italy were: age 40–80 years, IPF diagnosed in past 48 months, FVC ≥50% of theoretical value and DLCO ≥35% of predicted value. Exclusion criteria for Pirfenidone treatment were hypersensitivity to the active substance or to any of the excipients, severe liver function or liver disease in terminal stage, severe renal impairment (CrCl <30ml/min) or kidney disease in terminal stage requiring dialysis. When the patients fulfilled criteria for use of both drugs (Nintedanib and Pirfenidone) the final decision was based on contraindications and patient choice when faced with the different side-effect profiles. A relevant aspect was the dosage (Pirfenidone is administered as three capsules taken three times a day, whereas Nintedanib only one capsule twice a day).
All the clinical, radiological, immunological parameters were entered in a database and updated every 3 months during clinical assessment.
Statistical analysisAll data was expressed as mean±standard deviation. The Wilcoxon test was used to describe the trend of PFR patients in the two treatment groups. Analysis of variance (with Bonferroni test for internal comparisons) was used to compare groups. Statistical significance was set at p<0.05. Statistical analysis was carried out using StatSoft (2001) and Graph Pad Prism software.
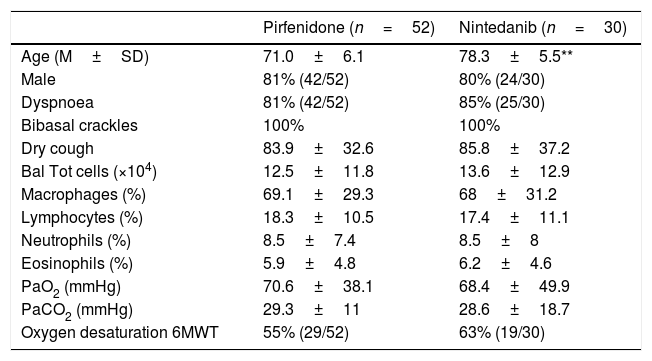
ResultsTable 1 shows the clinical characteristics of 82 IPF patients enrolled in this study in the last 12 months: 52 patients were treated with Pirfenidone and 30 patients with Nintedanib. The mean age of those treated with Pirfenidone was 71.04±6.1 years compared to 78.33±5.5 years of those treated with Nintedanib. The median age of patients treated with Nintedanib was significantly higher than that of patients in the other group (p<0.0001) due to the Pirfenidone group inclusion criteria (in Italy only patients under 80 years old can be treated with this drug while there is no age limit for Nintedanib). As expected, there was a clear prevalence of males in our IPF population (80%) with no difference between groups (Fig. 1). Smoking history revealed 74% of smokers in the Pirfenidone group and 52% in the Nintedanib group. Table 1 also shows that the most common symptoms at disease onset were exertional dyspnoea and dry cough, with no difference between groups. All patients manifested bibasal crackles during physical examination, and clubbing was evident in 35% of cases (Table 1).
Clinical features of IPF patients treated with Pirfenidone (n=52) or Nintedanib (n=30).
| Pirfenidone (n=52) | Nintedanib (n=30) | |
|---|---|---|
| Age (M±SD) | 71.0±6.1 | 78.3±5.5** |
| Male | 81% (42/52) | 80% (24/30) |
| Dyspnoea | 81% (42/52) | 85% (25/30) |
| Bibasal crackles | 100% | 100% |
| Dry cough | 83.9±32.6 | 85.8±37.2 |
| Bal Tot cells (×104) | 12.5±11.8 | 13.6±12.9 |
| Macrophages (%) | 69.1±29.3 | 68±31.2 |
| Lymphocytes (%) | 18.3±10.5 | 17.4±11.1 |
| Neutrophils (%) | 8.5±7.4 | 8.5±8 |
| Eosinophils (%) | 5.9±4.8 | 6.2±4.6 |
| PaO2 (mmHg) | 70.6±38.1 | 68.4±49.9 |
| PaCO2 (mmHg) | 29.3±11 | 28.6±18.7 |
| Oxygen desaturation 6MWT | 55% (29/52) | 63% (19/30) |
No significant differences were found between the two groups, except that mean age was significantly higher in the Nintedanib than the Pirfenidone group (**p<0.001).
BAL=bronchoalveolar lavage, M=macrophages, L=lymphocytes, N=neutrophils, E=eosinophils. The results are reported as medium±standard deviation.
 versus Nintedanib (80%).")
Fig. 2 indicates lung function test parameters at time 0 and after 6 and 12 months in the population of IPF patients treated with Pirfenidone. At time 0, FVC, FEV1, DLCO and TLC percentages were 78.7±16.9% predicted values, 83.3±16.5% predicted, 51.2±15.3% predicted and 68.7±14.7% predicted, respectively.
, after 6 months (n=35) and 12 months (n=25) in patients treated with Pirfenidone. No statistically significant differences among the groups were observed (p>0.05).")
Fig. 3 shows lung function test parameters at time 0 and after 6 and 12 months in the population of IPF patients treated with Nintedanib. At time 0, FVC, FEV1, DLCO and TLC percentages were 80.9±19.6% predicted, 87±19% predicted, 47±15% predicted and 66.3±13% predicted, respectively. No significant differences in FVC, FEV1, TLC and DLCO percentages were found at time 0 between patients treated with the two drugs (p=0.59, p=0.37, p=0.21, p=0.48, respectively).
, after 6 months (n=16) in patients treated with Nintedanib. No statistical significant differences among these groups were observed (p>0.05).")
At 6-month follow-up, FVC, FEV1, DLCO and TLC percentages of predicted values were 83±17.3% predicted, 88.6±18.3% predicted, 50.7±13.3% predicted and 71.3±15% predicted, respectively, in patients treated with Pirfenidone, and 78.3±18% predicted, 84.3±17.8% predicted, 47.6±15.3% predicted and 67.15±16% predicted, respectively, in patients treated with Nintedanib (Figs. 2 and 3). Interestingly, no significant differences in FVC, FEV1, DLCO or TLC % of predicted values were found between patients treated with Pirfenidone and Nintedanib at 6-month follow-up (p=0.54, p=0.38, p=0.76, p=0.31, respectively).
Only IPF patients treated with Pirfenidone reached 12-month follow-up because Nintedanib was approved in Italy more recently and only a limited number of patients treated with this drug had completed 1 year of treatment at the time of writing. At 12-month follow-up, FVC, FEV1, TLC and DLCO % of predicted values of IPF patients treated with Pirfenidone were 81.2±20.7% predicted, 86.7±19.7% predicted, 70.2±19.2% predicted and 46.7±9.8% predicted, respectively. No statistical difference in any lung function test parameter was observed between time 0 and 6 or 12 months in the Pirfenidone therapy group (p>0.05) (Figs. 2 and 3).
Pirfenidone showed a good tolerability profile in the majority of IPF patients. It was manageable and the most frequent side effects included gastrointestinal disorders and skin reactions. In our population dyspepsia and nausea were the most frequent adverse event occurring in 35% of cases followed by diarrhea (28% of patients) and cutaneous rash (19% of cases). The use of prokinetic drugs and proton-pump inhibitors allowed to improve gastrointestinal side effects in the majority of patients as well as the application of a broad-spectrum sunscreen. In three patients the rush and the photosensitivity were severe consequent to the persistent sun exposure for work reasons.
Nintedanib therapy was well tolerated. The most frequent side effect was diarrohea occurring in 40% of patients and requiring in 18% of cases a premature discontinuation. Treatment interruption was necessary only in 5% of patients, the other subjects improved after dose reduction from 150 to 100mg twice a day. 8% of patients treated with this drug presented altered liver enzymes, reversible after dose reduction. In four patients the elevation of aminotransferase concentrations tended to persist after six months of treatment and liver alteration were not reversible with dose modification and discontinuation. Both these patients had a persisting mild liver dysfunction secondary to lithiasis.
DiscussionSafety and efficacy outcomes of Pirfenidone and Nintedanib treatment in patients with IPF are available from many international clinical trials.7,10–13 Very little data is currently available about real-life experience with antifibrotic drugs in IPF patients.15–17 The present study evaluated the effectiveness of Pirfenidone and Nintedanib therapy in a population of IPF patients diagnosed in the last 12 months at Florence ILD Referral Centre. This observational study offers information on drug effectiveness in the real clinical setting of a university hospital, but can only partly be compared with international clinical trials and it has the limit that did not show 12 months follow-up for patients treated with Nintedanib. As expected, there was a high prevalence of male over female patients: while ILDs associated with connective tissue disorders are prevalent in women, IPF is typical of males over 65 years of age.18 The patients in the Nintedanib subgroup were older than those in the Pirfenidone group, because in Italy IPF patients over 80 years old can only be treated with Nintedanib and not with Pirfenidone.
In line with the literature, all patients (100%) presented bibasal crackles and restrictive functional deficit with a significant reduction in DLCO at disease onset.19,20 At 6-month follow-up both drugs appeared to stabilize the spirometric profile of a disease typically associated with deterioration of lung function in the period of a few months. In the Pirfenidone group, stabilization was also confirmed at 12-month follow-up, in line with reports on the literature. In our real-experience study, irrespective of baseline functional status, treatment with the two drugs was associated with functional benefits that were similar to those reported in clinical trials9 and with similar rates of FVC decline. Stabilization of lung function parameters was also observed in the older population of patients treated with Nintedanib.
This preliminary study assessing the therapeutic benefit of Pirfenidone and Nintedanib in an Italian cohort of IPF patients revealed a stabilization of lung function test parameters. The antifibrotic drugs also appeared to provide functional stabilization in older IPF/UIP patients.
Compliance with ethical standardsThis study was unfunded. None of the authors have a conflict of interest to declare.
Ethical approval: All procedures were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with animals performed by any of the authors.
Informed consent: Informed consent was obtained from all individual participants included in the study.
Conflict of interestThe authors have no conflicts of interest to declare.
The following are the supplementary data to this article:


