




Chronic hypersensitivity pneumonitis (CHP) is an interstitial lung disease with limited treatment response and bad prognosis. Sometimes it is indistinguishable from idiopathic pulmonary fibrosis (IPF) becoming one of the main differential diagnosis. The aim of our study is to compare survival and functional decline between these two entities.
MethodsSurvival and functional decline more than 10% in FVC were compared using Kaplan Meier (KM) method between patients with CHP and IPF. Cox proportional hazard analysis was used to identify independent predictors of survival and functional decline.
Results146 patients were included, 54 with CHP and 92 with IPF. KM rate for 2 years survival was 0.71 (CI 95% 0,6–0,8) for CHP group and 0,83 (CI 95% 0,66 - 0,92) for IPF (p=0,027). Nevertheless this difference disappeared using Cox proportional hazard analysis, the adjusted HR for survival among CHP patients was 0,53 (CI 95% 0,25–1,15) (p=0,11). There was no difference in functional evolution between the two groups. KM rate for a decline more than or equal to 10% was 0,64 for CHP (CI 95% 0,43–0,79) and 0,78 for IPF (IC 95% 0,6–0,88) (p=0,22). This observation did not change after using Cox proportional hazard analysis.
ConclusionsOur study shows that both IPF and CHP are fibrosing interstitial diseases with a similar evolution and survival. It might be possible that therapeutic approach in patients with CHP should change in the light of these observations.
Hypersensitivity pneumonitis (HP) is a complex syndrome caused by the repeated inhalation of antigenic particles, most of them organic, found in different environments. In susceptible patients, these antigens cause an exaggerated immune response compromising small airways and lung parenchyma.1 Due to the lack of validated diagnostic criteria, the real prevalence and incidence are unknown, contributing to the underdiagnosis of HP.
Traditionally, HP was classified as acute, subacute and chronic based on symptoms, tomographic and histopathologic findings, but because of significant overlap between these categories, some authors have currently proposed to classify HP as chronic – fibrotic and acute – inflammatory.2–4 The suspicion of HP should always be present in the diagnostic workup of a patient with interstitial lung disease, especially in those with chronic pattern. Sometimes, the diagnosis of HP can be challenging due to the similarity to idiopathic pulmonary fibrosis (IPF) and contributing to the misdiagnosis of the disease.5,6 Patients with tomographic and/or histological chronic-fibrotic findings have worse prognosis and poor treatment response to immunosuppressive therapy.7–9 Interestingly, some observational studies suggest that CHP might have better survival than IPF, although, there is no definitive evidence.9,10
The aim of our study is to compare survival between patients with CHP and IPF. The secondary objective is to compare respiratory function decline between these two entities.
MethodsWe conducted a retrospective cohort study. Patients with CHP and IPF followed up in a dedicated interstitial lung disease service were included between March 2012 and May 2017. Patients gave their informed consent to participate in the study. For IPF diagnosis, 2011 guideline criteria were used.11 A multidisciplinary discussion in conjunction with a modified algorithm (proposed by referred authors) was used to diagnose CHP (Fig. 1).1,3 The diagnosis of CHP was made if patients had positive findings in at least two of the three following domains: 1) High resolution CT scan (HRCT), 2) Cytology/Histology [bronchoalveolar lavage (BAL) and/or transbronchial biopsy (TBB)] or surgical biopsy, 3) Exposure to a potential antigen detailed by the patient during the clinical interview. CHP diagnosis was also considered when compatible findings were present in specimens of surgical lung biopsy alone, but, only after multidisciplinary discussion.
 centrilobular nodules. - High risk patients: cardiovascular comorbidities to perform invasive procedures and/or low FVC and DLCO/rest hypoxemia/patient’s refusal to do the procedure. High risk patients with a HCRT not compatible with CHP and/or without antigen were considered unclassifiable ILD and were excluded from this algorithm. - Classical or relevant antigen: Avian antigen and household moulds. HRCT: High resolution CT scan; ILD: interstitial lung disease; CHP: Chronic hypersensitivity pneumonitis; BAL: bronchoalveolar lavage; TBB: transbronchial biopsy; Ly: lymphocytosis; MDD: multidisciplinary discussion.")
Diagnostic algorithm for Chronic hypersensitivity pneumonitis.
*Bronchiectasis and traction bronchiolectasis and/or reticulation and/or honeycombing+bronchiolocentric, upper and middle lobe distribution of the abnormalities and/or mosaic pattern and/or ground glass (GG) centrilobular nodules.
- High risk patients: cardiovascular comorbidities to perform invasive procedures and/or low FVC and DLCO/rest hypoxemia/patient’s refusal to do the procedure. High risk patients with a HCRT not compatible with CHP and/or without antigen were considered unclassifiable ILD and were excluded from this algorithm.
- Classical or relevant antigen: Avian antigen and household moulds.
HRCT: High resolution CT scan; ILD: interstitial lung disease; CHP: Chronic hypersensitivity pneumonitis; BAL: bronchoalveolar lavage; TBB: transbronchial biopsy; Ly: lymphocytosis; MDD: multidisciplinary discussion.
The following variables were described: age, sex, tobacco exposure, gastroesophageal reflux disease (GERD) symptoms, dyspnoea severity (assessed with the mMRC scale), digital clubbing. Pulmonary function tests (PFTs), six minutes walking test (6 MWT), high resolution CT (HRCT), and histology/cytology data were also recorded.
Variable definitionsPulmonary function tests: spirometry, lung volumes (measured with pletismography), diffusion capacity (expressed as DLCO) and 6 MWT were performed at the time of diagnosis and every 4–6 months according to reference procedures guidelines.12–17
HRCT: performed in all the patients and reported by a radiologist specialized in ILD. It was classified as typical, possible or inconsistent with UIP.11 HRCT findings considered compatible with HP were: mosaic pattern, ground glass (GG) centrilobular nodules, with and/or bronchiolocentric, and upper and middle lobe distribution.3,18,19 The presence of bronchiectasis, traction bronchiolectasis, reticulation and honeycombing were considered compatible with a chronic form of HP.
Lung biopsy: it was reported by an experienced pathologist specialized in ILD. IPF diagnosis was done according to 2011 guidelines.11 HP diagnosis was based on the presence of poorly formed granulomas, giant cells, bronchiolocentric interstitial pneumonia, nonspecific interstitial pneumonia (NSIP), organizing pneumonia, centrilobular fibrosis and bridging fibrosis. Also, UIP pattern, fibrotic NSIP, honeycombing and interstitial fibrosis were considered to define CHP.
Statistical analysisResults were expressed as mean and standard deviation (SD), or median and interquartile range (IQR), if variables were continuous and as absolute frequency or percentage if variables were categorical. Chi square, Fisher´s exact test, Mann Whitney test or t student test were used for comparisons.
Time to death and time to a decline of 10% or more in the percentage of the predicted forced vital capacity (FVC%) from the basal measurement were estimated using Kaplan–Meier methods. The decline in FVC was calculated by considering the difference between percentages of predicted values. The rates of events between groups at 24 months were studied using survival estimators and 95% confidence intervals, and compared using the log rank test. A multivariate Cox model was applied, using HP as the first independent variable. Variables that were significant in the univariate analysis and those considered clinically relevant were included. Both adjusted and unadjusted HR, and their 95% CI, were reported. A p-value of <0,05 was considered statistically significant.
We calculated the statistical power for the difference in survival between patients with CHP and IPF. Considering a fixed sample size of 146 patients, a hypothesis with two tails, and an estimated HR of 0,6, a statistical power of 0,86 was obtained.
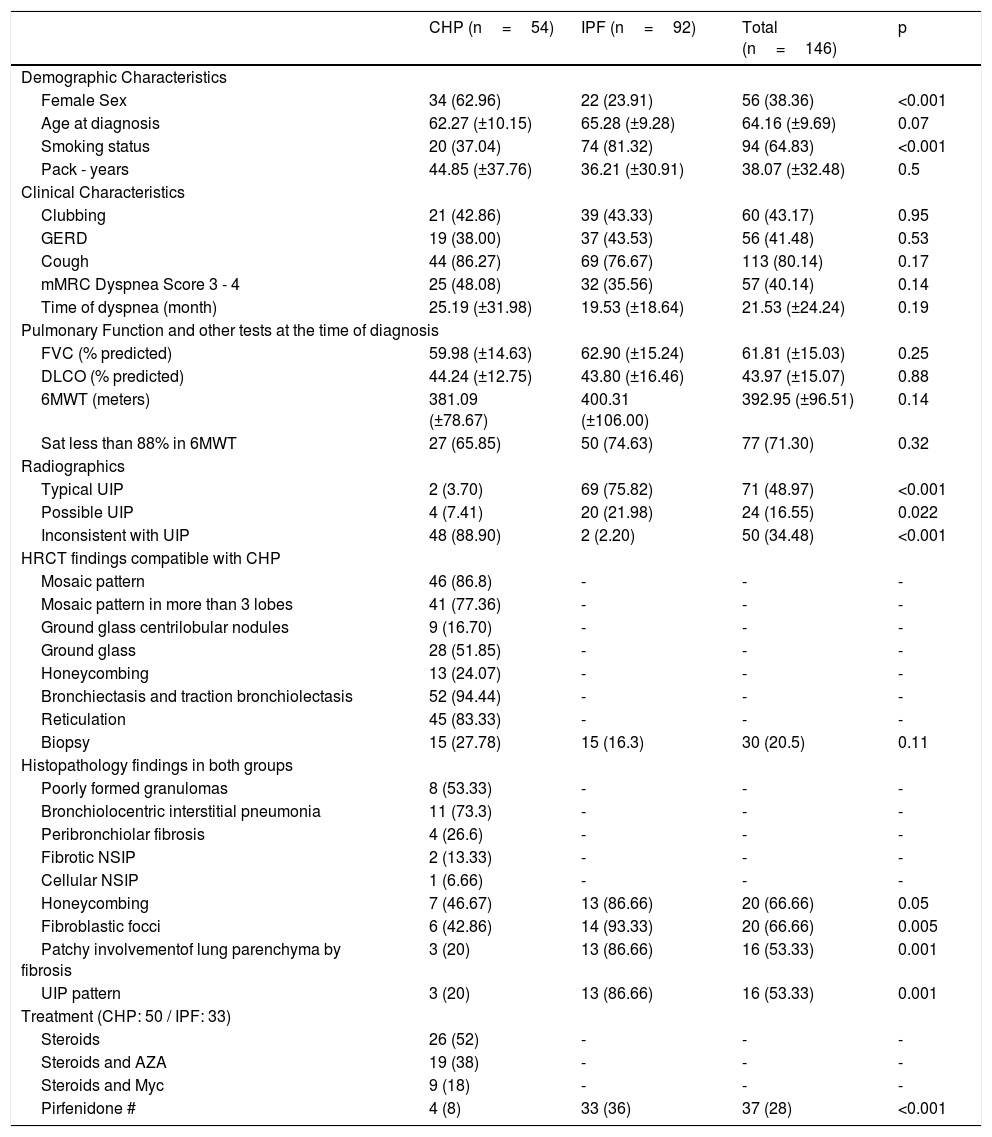
Results146 patients were included; 54 (37%) with CHP and 92 (63%) with IPF. Cohort characteristics and comparison between groups are shown in Table 1. Patients within the CHP group were more likely female (62,96% vs. 23,91%, p<0,001) and less likely smokers (37,04% vs 81,32%, p<0,001) in comparison with the IPF group. At the time of diagnosis, there were no statistically significant differences between groups (CHP/IPF) in terms of: age, dyspnoea (length and severity) PFTs results and 6 MWT.
Patient cohort with CHP and IPF. Description of demographic, clinical and tomographic variables. Comparison of both cohorts
| CHP (n=54) | IPF (n=92) | Total (n=146) | p | |
|---|---|---|---|---|
| Demographic Characteristics | ||||
| Female Sex | 34 (62.96) | 22 (23.91) | 56 (38.36) | <0.001 |
| Age at diagnosis | 62.27 (±10.15) | 65.28 (±9.28) | 64.16 (±9.69) | 0.07 |
| Smoking status | 20 (37.04) | 74 (81.32) | 94 (64.83) | <0.001 |
| Pack - years | 44.85 (±37.76) | 36.21 (±30.91) | 38.07 (±32.48) | 0.5 |
| Clinical Characteristics | ||||
| Clubbing | 21 (42.86) | 39 (43.33) | 60 (43.17) | 0.95 |
| GERD | 19 (38.00) | 37 (43.53) | 56 (41.48) | 0.53 |
| Cough | 44 (86.27) | 69 (76.67) | 113 (80.14) | 0.17 |
| mMRC Dyspnea Score 3 - 4 | 25 (48.08) | 32 (35.56) | 57 (40.14) | 0.14 |
| Time of dyspnea (month) | 25.19 (±31.98) | 19.53 (±18.64) | 21.53 (±24.24) | 0.19 |
| Pulmonary Function and other tests at the time of diagnosis | ||||
| FVC (% predicted) | 59.98 (±14.63) | 62.90 (±15.24) | 61.81 (±15.03) | 0.25 |
| DLCO (% predicted) | 44.24 (±12.75) | 43.80 (±16.46) | 43.97 (±15.07) | 0.88 |
| 6MWT (meters) | 381.09 (±78.67) | 400.31 (±106.00) | 392.95 (±96.51) | 0.14 |
| Sat less than 88% in 6MWT | 27 (65.85) | 50 (74.63) | 77 (71.30) | 0.32 |
| Radiographics | ||||
| Typical UIP | 2 (3.70) | 69 (75.82) | 71 (48.97) | <0.001 |
| Possible UIP | 4 (7.41) | 20 (21.98) | 24 (16.55) | 0.022 |
| Inconsistent with UIP | 48 (88.90) | 2 (2.20) | 50 (34.48) | <0.001 |
| HRCT findings compatible with CHP | ||||
| Mosaic pattern | 46 (86.8) | - | - | - |
| Mosaic pattern in more than 3 lobes | 41 (77.36) | - | - | - |
| Ground glass centrilobular nodules | 9 (16.70) | - | - | - |
| Ground glass | 28 (51.85) | - | - | - |
| Honeycombing | 13 (24.07) | - | - | - |
| Bronchiectasis and traction bronchiolectasis | 52 (94.44) | - | - | - |
| Reticulation | 45 (83.33) | - | - | - |
| Biopsy | 15 (27.78) | 15 (16.3) | 30 (20.5) | 0.11 |
| Histopathology findings in both groups | ||||
| Poorly formed granulomas | 8 (53.33) | - | - | - |
| Bronchiolocentric interstitial pneumonia | 11 (73.3) | - | - | - |
| Peribronchiolar fibrosis | 4 (26.6) | - | - | - |
| Fibrotic NSIP | 2 (13.33) | - | - | - |
| Cellular NSIP | 1 (6.66) | - | - | - |
| Honeycombing | 7 (46.67) | 13 (86.66) | 20 (66.66) | 0.05 |
| Fibroblastic focci | 6 (42.86) | 14 (93.33) | 20 (66.66) | 0.005 |
| Patchy involvementof lung parenchyma by fibrosis | 3 (20) | 13 (86.66) | 16 (53.33) | 0.001 |
| UIP pattern | 3 (20) | 13 (86.66) | 16 (53.33) | 0.001 |
| Treatment (CHP: 50 / IPF: 33) | ||||
| Steroids | 26 (52) | - | - | - |
| Steroids and AZA | 19 (38) | - | - | - |
| Steroids and Myc | 9 (18) | - | - | - |
| Pirfenidone # | 4 (8) | 33 (36) | 37 (28) | <0.001 |
CHP: Chronic hypersensitivity pneumonitis; IPF: idiopathic pulmonary fibrosis; GERD: gastroesophageal reflux disease; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide; 6MWT: six minutes walking test; Sat: oxygen saturation; HRCT: High resolution computed tomography; UIP: usual interstitial pneumonia, NSIP: nonspecific interstitial pneumonia, AZA: azathioprine, Myc: Mycophenolate.
Data expressed as n (%) or mean (SD). # p<0,05 for the comparison between CHP and IPF.
Patients with IPF had a higher frequency of HRCT with typical or possible UIP pattern. In contrast, patients with CHP had a higher proportion of inconsistency with UIP pattern. The most frequent HRCT findings compatible with HP were: mosaic pattern (86.8%), mosaic pattern in more than three lobes (77.36%) and ground glass centrilobular nodules (16.7%).
Thirty surgical biopsies were performed, 27.7% in the CHP group, and 16.67% in the IPF group. In the former, poorly formed granulomas were found in 8 samples, while bronchiolocentric interstitial pneumonia and NSIP were described in 11 and 3 biopsies, respectively.
Fifty patients with CHP were treated. In all these cases, steroid therapy was used; 26 (52%) as monotherapy, 19 (38%) in conjunction with mycophenolate and 9 (18%) with azathioprine (four patients received both immunosuppressants). Pirfenidone was used in four patients (8%). 33 (36%) patients with IPF received antifibrotic treatment with pirfenidone (Table 1).
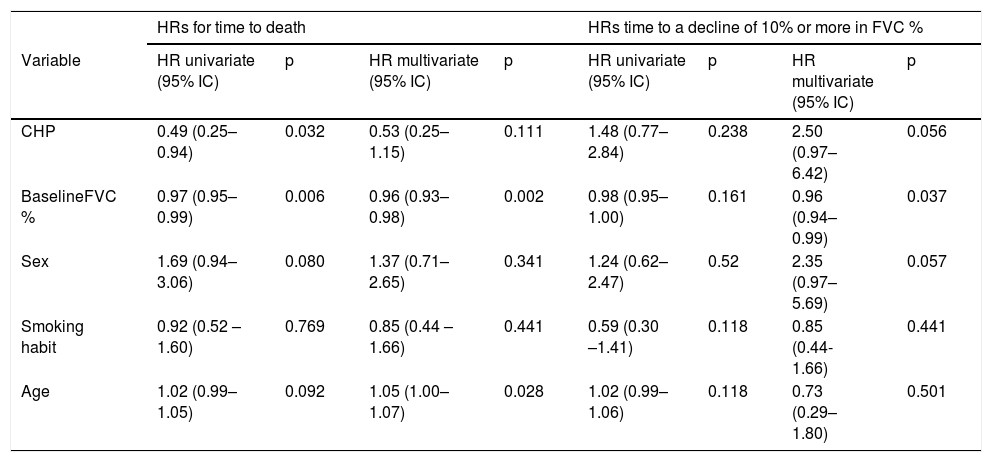
Survival analysisAll the patients were included for the survival analysis. Forty-two deaths were observed among IPF patients, and twelve in the CHP group. The KM estimator for 2-year survival was 0,71 (95% CI 0,6–0,8) in the IPF group and 0,83 (95% CI 0,66–0,92) in the CHP group; the difference between both was statistically significant (p=0,027) (Fig. 2). Using Cox univariate analysis CHP diagnosis and FVC% at the time of diagnosis were associated with better survival. In the multivariate analysis, patients with higher FVC% and younger age at the time of diagnosis showed better survival rates, and the effect of the diagnosis of CHP on survival disappeared. (Table 2)
. CHP: Chronic hypersensitivity pneumonitis; IPF: idiopathic pulmonary fibrosis.")
Univariate and multivariate HRs for time to death [cohort of patients with CHP and IPF (N=146)] and time to a decline of 10% or more in FVC% [cohort of patients with CHP and IPF (n=91)].
| HRs for time to death | HRs time to a decline of 10% or more in FVC % | |||||||
|---|---|---|---|---|---|---|---|---|
| Variable | HR univariate (95% IC) | p | HR multivariate (95% IC) | p | HR univariate (95% IC) | p | HR multivariate (95% IC) | p |
| CHP | 0.49 (0.25–0.94) | 0.032 | 0.53 (0.25–1.15) | 0.111 | 1.48 (0.77–2.84) | 0.238 | 2.50 (0.97–6.42) | 0.056 |
| BaselineFVC % | 0.97 (0.95– 0.99) | 0.006 | 0.96 (0.93–0.98) | 0.002 | 0.98 (0.95–1.00) | 0.161 | 0.96 (0.94–0.99) | 0.037 |
| Sex | 1.69 (0.94– 3.06) | 0.080 | 1.37 (0.71– 2.65) | 0.341 | 1.24 (0.62–2.47) | 0.52 | 2.35 (0.97–5.69) | 0.057 |
| Smoking habit | 0.92 (0.52 – 1.60) | 0.769 | 0.85 (0.44 – 1.66) | 0.441 | 0.59 (0.30 –1.41) | 0.118 | 0.85 (0.44-1.66) | 0.441 |
| Age | 1.02 (0.99– 1.05) | 0.092 | 1.05 (1.00–1.07) | 0.028 | 1.02 (0.99–1.06) | 0.118 | 0.73 (0.29–1.80) | 0.501 |
CHP: Chronic hypersensitivity pneumonitis; IPF: idiopathic pulmonary fibrosis; FVC%: forced vital capacity.
Ninety-one patients were included in this analysis, 36 with CHP diagnosis and 55 with IPF diagnosis. The frequency of the event was 16 among patients with CHP and 23 in those with IPF. The KM estimator was 0,78 (95% CI 0,6–0,88) in the IPF group and 0,64 (95% CI 0,43–0,79) in the CHP group. No statistically significant difference was found between the two groups (p=0,22) (Fig. 3). By Cox univariate analysis higher FVC% and a younger age at diagnosis were associated with a lesser FVC% decline over time. These associations remained in the multivariate analysis (Table 2).
Discussion
In our cohort we observed that patients with CHP had a similar survival at two years compared to patients with IPF. The difference in survival observed in the univariate analysis related to the diagnosis of CHP disappeared in the multivariate analysis. On the other hand, higher baseline percentage of the predicted FVC remained as a better prognostic factor in the multivariate analysis. These results agree with previous studies, where patients with CHP and fibrotic histologic pattern (UIP, fibrotic NSIP and peribronchiolar fibrosis) or the presence of honeycombing in HRCT had a similar survival rate to those with IPF.20–23 On the other hand our results are discordant with a study where patients with CHP had survived better than patients with IPF, despite the groups having a similar fibrosis score.24
Moreover, we did not find any statistically significant difference in FVC% decline between the groups. In a retrospective study, a decline of ≥10% in FVC% after 6– 12 months was associated with a reduction in survival in CHP, irrespective of the initial FVC% value, behaving like the IPF group.25 Greater values of FVC% at baseline and a younger age at the time of diagnosis were factors associated with better survival and a lesser rate of decline in FVC% for both groups. These results support what was previously reported by Ryerson et al. and Ley et al.9,26 In line with these findings, a recent study found that older age, low percentage of lymphocytes in BAL, decreased DLCO, honeycombing in HRCT and the presence of UIP pattern were factors associated with lower survival rates for patients with CHP.27
Fewer patients diagnosed with CHP were smokers or male sex, similar to that described in other studies.24,25,28 This information could be useful to differentiate the two entities, helping with the diagnosis dilemma in daily practice.
Since the initial injury mechanism of HP is characterized by inflammation, good response to immunotherapy should be expected. However, in our study we did not find statistically significant differences in survival or respiratory function decline between patients with IPF and those with CHP, suggesting a lack of effectiveness of immunosuppressive treatment in the latter. This observation is in accordance with a recent study where corticosteroid treatment was not effective in patients with fibrotic HP (according to HRCT) regarding FVC% and DLCO%, and whether patients received steroid treatment or not made no difference to survival rates.29 Moreover, no improvement in FVC% was seen in patients who received treatment with azathioprine or mycophenolate.30 In a retrospective study, the use of mycophenolate or azathioprine reduced the frequency of adverse effects compared to steroids as monotherapy, without benefits in terms of respiratory function decline or survival. Interestingly, the same study showed that patients diagnosed with CHP who did not received immunosuppressive treatment with steroids either as monotherapy, or associated with mycophenolate or azathioprine, survived longer.31 These results suggest that the use of immunosuppressants in patients with predominantly fibrotic HP could be harmful, as has been previously described in IPF patients.32
In recent years, the efficacy and safety of antifibrotic drugs (nintedanib and pirfenidone) in the treatment of patients with IPF have been demonstrated.33,34 In this scenario, the use of these drugs in predominantly fibrotic interstitial pathology of other etiologies appears as a reasonable alternative. This is the case of CHP, in which clinical trials are being conducted to study this hypothesis (www.ClinicalTrials.gov identifier NCT02496182 and NCT02999178).35,36 It is possible that in a near future the treatment of the chronic-fibrotic phases of HP will be based on the combination of an immunosuppressant and an antifibrotic drug.
In our cohort less than half of the patients with IPF received antifibrotic treatment with pirfenidone and this is explained by several reasons. Since the approval of pirfenidone and nintedanib, not all patients with IPF in our country can get access to these drugs or they have to wait for a long time until their health care provider approves the medication. To support this, it was observed, in a real life study done in our centre, that the mean (SD) of days between the diagnosis of IPF and the start of treatment with pirfenidone was 28,340 days (SD 2017). We also found that the most frequent reason for discontinuation was failure of suppliers to provide the drug 37. In our cohort, four patients with CHP received pirfenidone as an “off label” indication. This was supported by our multidisciplinary team in view of the lack of efficacy of just the immunosuppressive treatment.
Our study has some limitations. First, since there is not a gold standard for the diagnosis of HP, we used an adapted version of the algorithm proposed by other authors. This could make comparison of our results with those of other studies that have used other criteria controversial. Secondly, we should have used a fibrosis score in HRCT as an adjustment variable in the multivariate analysis. Because of the impossibility of obtaining such a score, given the retrospective nature of the study, we used FVC% at the time of diagnosis as a surrogate. Finally, we have all the inherent limitations of a retrospective study, such as the possible presence of a selection bias. Despite the limitations mentioned above, we believe that our results can be extrapolated, and contribute to the current knowledge of these diseases.
In conclusion, patients with CHP have a similar rate of FVC% decline and survival to patients with IPF, which highlights the importance of establishing an early diagnosis, and raises the need to study new therapeutic options for this entity.
Dr. Santiago Giavedoni.


