

Cystic adenomatoid malformation (CAM) of the lung is thought to be a hamartomatous lesion, characterized by bronchiolar maturation disturbance,1,2 it is very rare in adults3 and pathogenesis is still unknown.4 CAM is classified according to Yousem et al.5 into 4 types which are related with the underdeveloped bronchial-pulmonary segment and histological features. According to the literature the prevalence is as follows: type 1 (50–70%), type 2 (20–40%), type 4 (10–15%), type 3 (10%) and type 0 (1–3%). Clinical presentation varies (perinatal death, repeat infections, hemoptysis to lung carcinomas6) and severity relates to the size of the lesion; it is more frequent in the lower lobes and rarely affects more than one lobe. Chest radiograph reveals rounded thin-walled spaces, filled with air or secretions, although CT scan correlates with better morphological characterization. The mainstay treatment is pulmonary resection, usually due to the risk of complications such as infection, cystic expansion or malignant transformation.6 For asymptomatic patients some authors recommend a conservative approach.
In order to get a better characterization of CAM in adults in our center, all cases between 2008 and 2015 were reviewed. CAM was diagnosed in 17 patients admitted to Coimbra Hospital and University Centre (CHUC). Patient demographics, presenting symptoms, chest radiography, CT scan, type of surgery and histopathology result were analyzed.
Five males and twelve females, aged between 20 and 85 years, with a mean age of 53.4 years were reviewed: the symptoms included recurrent infections in six patients, persistent productive cough in eight patients and hemoptysis in three. Chest radiographs and CT scans showed a pattern of bronchiectasis in seven patients, cystic lesions in six patients and nodular lesions in four patients; eight patients had it in the right lower lobe, four in the left lower lobe, two in the left upper lobe, one in the middle right lobe and two in more than one lobe in the right lung. All patients underwent surgery: lobectomy (9 cases) and pulmonary resection (8 cases). Histopathology reported two patients had CAM type 0, eight patients had CAM type 1, four patients had CAM type 2, one patient had CAM type 3 and CAM type 4 was not observed.
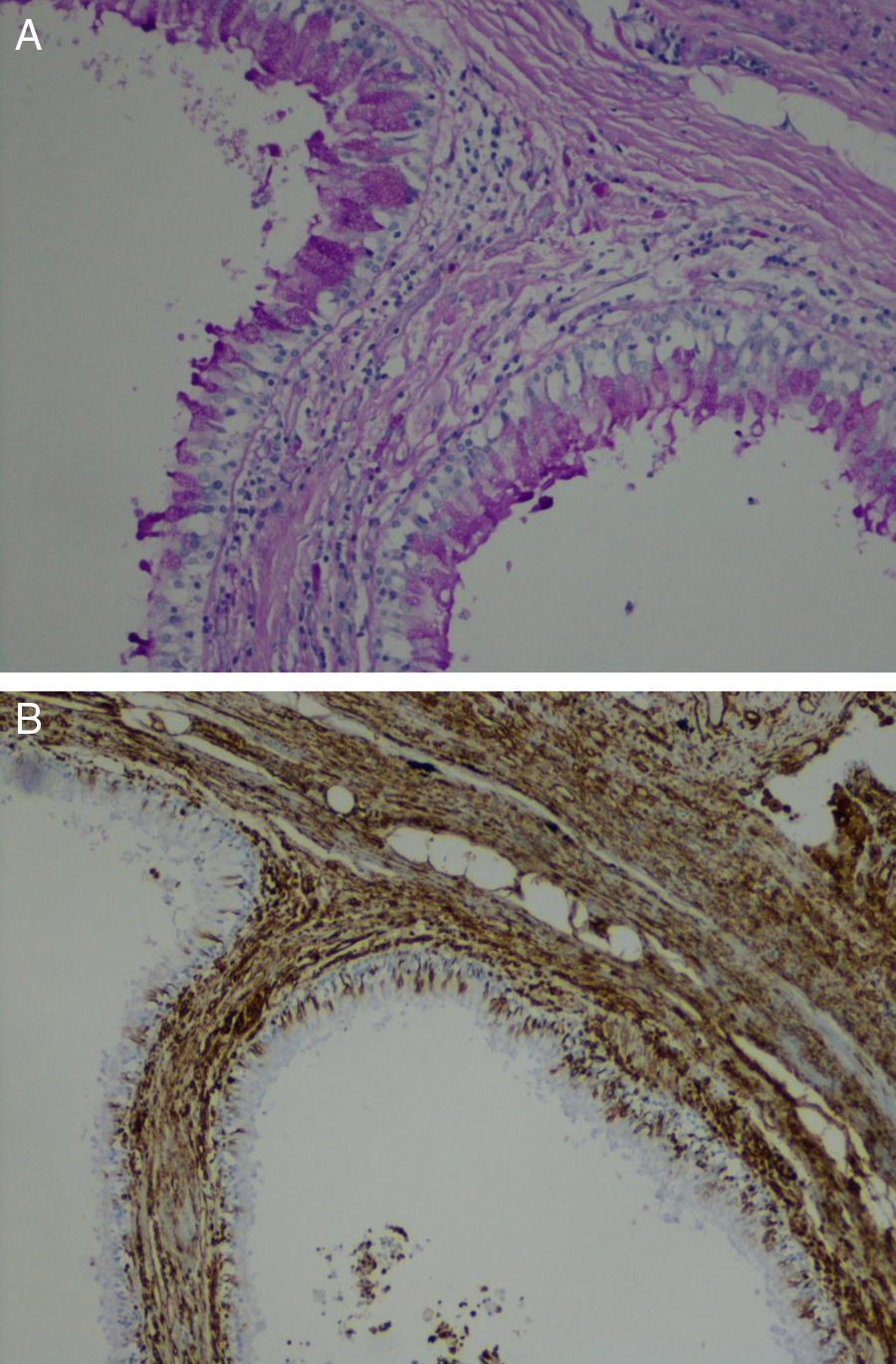
Our retrospective study included 17 adult patients with CAM. Although descriptions regarding gender differentials are not available, the majority of our patients were female. The most frequent complaints were recurrent infections and persistent productive cough, easily explained by the presence of bronchiectasis and fluid-filled cystic lesions depicted imagiologically. In most of these cases the presenting lesion was located in lower lobes, known from the literature to be the most common sites. The definitive diagnosis was only made by histological examination. The differential diagnosis included bronchogenic cysts, pulmonary sequestration, congenital lobar emphysema and mediastinal masses. According to the classification of Yousem et al.5 the CAM type 0 presents a tracheal/bronchial origin and involve all lobes. CAM type 1 is usually confined to a lobe with single or multiple cavitary lesions, with symptoms presenting in the first week of life.6 CAM type 2 is diagnosed in the first year of life and is associated with other malformations.6 CAM type 3 may involve the entire lung, usually without large space lesions.6 CAM type 4 has a distal acinar source and larger lesions that may develop pneumonia or pneumothorax; usually the diagnosis is incidental.7 Youssem et al.5 classification is a morphological compromise with prognostic value.7 Our series presented CAM type 1 as the most frequent type, in accordance with case series in the literature. CAM type 2 cases were not associated with other malformations. There was only one case of CAM type 3 affecting just the upper left lobe. There were no cases of CAM type 4 and there were two rare cases of CAM type 0 (Fig. 1). These two rare cases had neither symptoms nor history of infections in childhood and the location was atypical since it did not involve all the lobes and there were no associated lung malformations which could explain the absence of symptoms during childhood/adolescence.
 Irregular bronchial-like structures lined by pseudostratified ciliated columnar epithelium (PAS, ×400). (B) Columnar epithelium surrounded by prominent loose mesenchymal tissue (Vim, ×400).")
In conclusion, surgery is the gold standard treatment for CAM adult cases and for diagnosis by histopathology analysis. This study demonstrated how CAM with late clinical alterations onset is rare and difficult to diagnose, as most cases presented as incidental radiological findings.
FundingAbsence of financial support and off-label or investigational use.
Conflicts of interestThe authors have no conflicts of interest to declare.


