

Connective tissue diseases (CTD) are frequently associated with interstitial lung disease (ILD), significantly impacting their morbidity and mortality.
AimAnalyze the experience of an autoimmune specialized unit on treating CTD-ILD and characterize the population based on most frequent diseases, imaging patterns, lung function tests results, serology and treatment. Assess mortality and mortality predictors in these patients.
MethodsRetrospective, descriptive and statistical analysis of the CTD-ILD patients followed up at an autoimmune diseases unit during a 6-year period.
ResultsOver the study period, 75 patients with CTD-ILD were treated with a mean follow-up of 49 ± 31 months.
The most frequent CTD were systemic sclerosis and rheumatoid arthritis. ILD was diagnosed prior to CTD in 8% of patients and concomitantly in 35%. Nonspecific interstitial pneumonia was the CT pattern in 60% and 35% had an isolated diminished DLCO on lung function tests. Pulmonary hypertension was present in 12% and it was the single most important mortality predictor (OR 14.41, p = 0.006). Corticosteroids are the mainstay of treatment but biologics were prescribed in 39% of the patients (mostly tocilizumab and rituximab). Two scleroderma patients were recently treated with nintedanib.
ConclusionsILD is a potential complication of every CTD and can impose a dramatic burden on these patients. The clinical relevance of ILD together with their early expression in the course of the disease underlines the importance of the presence of chest physicians in these units.
Connective tissue diseases (CTD) are a group of diseases with heterogeneous systemic features and immune-mediated multi-organ disfunction. The respiratory tract can be targeted in virtually every CTD and with a multitude of manifestations, bringing important implications to the diagnosis, follow-up, treatment and prognosis of these patients.1,2 Lung involvement is a decisive contributor to the mortality in CTD, and is now the leading cause of death in systemic sclerosis (SSc)3 and an increasing cause of death in rheumatoid arthritis (RA), even as the overall mortality rates are falling.4
Interstitial lung disease (ILD) is frequently present in patients with autoimmune myopathies, SSc, Sjögren’s syndrome, RA and systemic lupus erythematosus (SLE), with estimated prevalences of 40%, 30–40%, 40%, 10% and 12%, respectively.5 It is important to keep in mind that ILD may be the only manifestation of a yet-to-be diagnosed CTD.6
High-resolution computed tomography (HRCT) and pulmonary function tests (PFT) are the best tools to evaluate lung involvement and have prognostic value.7–9
While there are no definitive serum biomarkers for CTD-ILD, the presence of some disease-specific markers can lead to a better screening and risk-stratifying of patients.10
Pulmonary hypertension (PH) is a common finding in CTD-ILD patients and adds significant impact to the morbidity and mortality of these patients.11
Development of lung disease, and its associated symptoms like dyspnea and cough, provides extra burden on a group of diseases already highly impactful on quality of life.12
Even though ILD is a common complication of CTD, the definitive guidance on how to treat these patients is scarce. Corticosteroids still represent the mainstay of treatment.13 In the context of SSc, there is also an increasing body of evidence for biologics, such as tocilizumab14 and rituximab,15 and renewed enthusiasm for new therapeutical targets, such as antifibrotic drugs.16,17
Due to the sheer complexity and systemic involvement of these diseases, a multidisciplinary approach should be the gold-standard when treating these patients.18 This paper aims to compile, analyze and discuss the experience of our unit in the management of CTD-ILD.
MethodsThe authors conducted a retrospective, descriptive analysis of patients older than 18 years diagnosed with CTD-ILD followed up at an autoimmune diseases outpatient clinic between January of 2013 and December of 2018.
The diagnosis of CTD was based on clinical and serologic criteria according to the most recent EULAR recommendations and the diagnosis of ILD was made on the basis of HRCT findings. All patients were discussed in a weekly autoimmune diseases multidisciplinary team meeting. Cases were further discussed with pulmonology and radiology on a case-by-case basis.
HRCT scans and PFT results shown are the ones done at the ILD diagnosis. HRCT scans were obtained with 1–1.5 mm thick slices. PFT were carried out according to a standardized protocol in the respiratory medicine department. Static lung volumes were measured using the plethysmography method, and the lung diffusion capacity of CO (DLCO) using the single breath-hold method. Antibodies were tested with the following techniques: ANA, ANCA – indirect immunofluorescence assay; Anti-SSA, anti-SSB, anti-Sm – Immunoblot assay; anti-dsDNA, anti-CCP – fluorescence enzyme immunoassay (FEIA); Rheumatoid factor – turbidimetry.
Data was collected from hospital records and handled in an anonymous and population-based fashion. Normally distributed variables are presented as mean and standard deviation and non-normal variables are presented as median and interquartile range. Differences in baseline variables were tested with t-tests for continuous variables; χ2-trend tests for ordered categorical variables; and χ2 tests for binomial and unordered categorical variables. Statistical analysis was performed using Stata® 14 software.
ResultsPopulationDuring the 6-year study period, 75 CTD-ILD patients were followed-up at this unit with a mean follow-up of 49 ± 31 months amounting to 646.2 patients-years. The mean age at the time of ILD diagnosis was 56 ± 15.5 years-old, there was a clear female predominance (77.3%) and the mean duration of CTD was 8.9 ± 7.5 years. CTD was diagnosed before ILD in 57% of patients, with a median interval of 49.1 [23.7–127.15] months separating the two diagnosis. ILD was diagnosed first in 8% of patients (n = 6) with CTD being diagnosed shortly after (mean interval of 5.1 months). Diagnosis was simultaneous in 35% of patients.
The CTD were, by order of prevalence, SSc (34.7%; n = 26), RA (20%; n = 15), overlap syndrome (13.3%; n = 10), mixed CTD (9.3%; n = 7), autoimmune myopathy (6.7%; n = 5), ANCA-positive vasculitis (5.3%; n = 4), undifferentiated CTD (5.3%; n = 4) and Sjögren syndrome (2.7%; n = 2).
Imaging and biopsyNonspecific interstitial pneumonia (NSIP) was the most common HRCT pattern, present in 60% of patients, followed by usual interstitial pneumonia (UIP) in 36% of patients and a pattern of lymphocytic interstitial pneumonia (LIP) in the remaining 3 patients. One patient with anti-synthetase syndrome had a predominance of NSIP pattern overlapped with organizing pneumonia (OP) features. Out of the patients with NSIP pattern, 42.2% had a diagnosis of SSc and 24.4% had overlap syndrome or mixed CTD with a predominance of scleroderma findings. Among patients with SSc, 58% had extensive disease (>20% involvement19) on HRCT. A LIP pattern was found in 2 patients with Sjögren syndrome and 1 with polymyositis. Other common findings were pleural thickening (n = 16), lung nodules (n = 14) and pleural effusion (n = 5).
While not mandatory for the diagnosis, lung biopsy was performed in 11 patients who had conflicting HRCT findings or a broader differential diagnosis, 3 being compatible with NSIP and 3 with UIP. The remaining 5 biopsies had nonspecific findings.
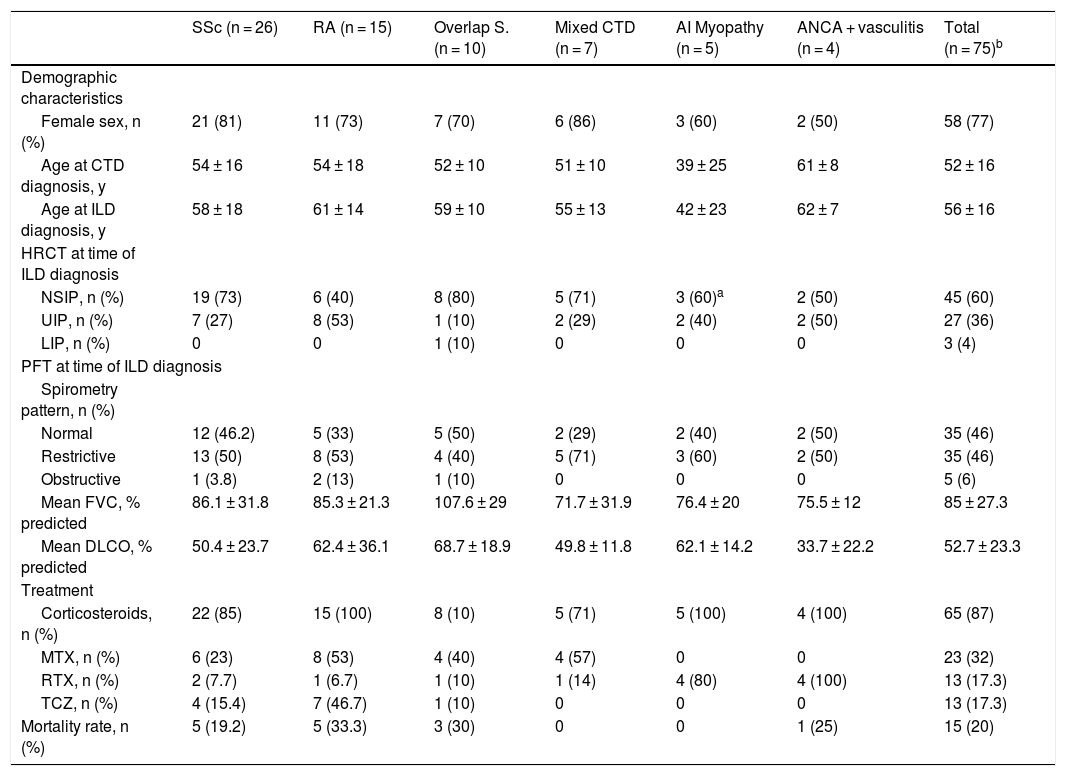
Pulmonary function testsPFT were carried out in all patients at the time of ILD diagnosis. A restrictive pattern was found in 46% whilst only 6% had an obstructive pattern. Isolated diminished DLCO was present in 35% of patients and 13% had all results within the normal range. ANCA-positive vasculitis, autoimmune myopathies and mixed CTD, were the subgroup of patients who presented with more severe lung involvement at diagnosis (Table 1).
Description of main characteristics analyzed by disease subgroups and the global cohort.
| SSc (n = 26) | RA (n = 15) | Overlap S. (n = 10) | Mixed CTD (n = 7) | AI Myopathy (n = 5) | ANCA + vasculitis (n = 4) | Total (n = 75)b | |
|---|---|---|---|---|---|---|---|
| Demographic characteristics | |||||||
| Female sex, n (%) | 21 (81) | 11 (73) | 7 (70) | 6 (86) | 3 (60) | 2 (50) | 58 (77) |
| Age at CTD diagnosis, y | 54 ± 16 | 54 ± 18 | 52 ± 10 | 51 ± 10 | 39 ± 25 | 61 ± 8 | 52 ± 16 |
| Age at ILD diagnosis, y | 58 ± 18 | 61 ± 14 | 59 ± 10 | 55 ± 13 | 42 ± 23 | 62 ± 7 | 56 ± 16 |
| HRCT at time of ILD diagnosis | |||||||
| NSIP, n (%) | 19 (73) | 6 (40) | 8 (80) | 5 (71) | 3 (60)a | 2 (50) | 45 (60) |
| UIP, n (%) | 7 (27) | 8 (53) | 1 (10) | 2 (29) | 2 (40) | 2 (50) | 27 (36) |
| LIP, n (%) | 0 | 0 | 1 (10) | 0 | 0 | 0 | 3 (4) |
| PFT at time of ILD diagnosis | |||||||
| Spirometry pattern, n (%) | |||||||
| Normal | 12 (46.2) | 5 (33) | 5 (50) | 2 (29) | 2 (40) | 2 (50) | 35 (46) |
| Restrictive | 13 (50) | 8 (53) | 4 (40) | 5 (71) | 3 (60) | 2 (50) | 35 (46) |
| Obstructive | 1 (3.8) | 2 (13) | 1 (10) | 0 | 0 | 0 | 5 (6) |
| Mean FVC, % predicted | 86.1 ± 31.8 | 85.3 ± 21.3 | 107.6 ± 29 | 71.7 ± 31.9 | 76.4 ± 20 | 75.5 ± 12 | 85 ± 27.3 |
| Mean DLCO, % predicted | 50.4 ± 23.7 | 62.4 ± 36.1 | 68.7 ± 18.9 | 49.8 ± 11.8 | 62.1 ± 14.2 | 33.7 ± 22.2 | 52.7 ± 23.3 |
| Treatment | |||||||
| Corticosteroids, n (%) | 22 (85) | 15 (100) | 8 (10) | 5 (71) | 5 (100) | 4 (100) | 65 (87) |
| MTX, n (%) | 6 (23) | 8 (53) | 4 (40) | 4 (57) | 0 | 0 | 23 (32) |
| RTX, n (%) | 2 (7.7) | 1 (6.7) | 1 (10) | 1 (14) | 4 (80) | 4 (100) | 13 (17.3) |
| TCZ, n (%) | 4 (15.4) | 7 (46.7) | 1 (10) | 0 | 0 | 0 | 13 (17.3) |
| Mortality rate, n (%) | 5 (19.2) | 5 (33.3) | 3 (30) | 0 | 0 | 1 (25) | 15 (20) |
SSc - Systemic sclerosis; RA - Rheumatoid Arthritis; Overlap S. - Overlap syndrome; CTD - connective tissue disorder; AI myopathy - Autoimmune myopathy; ILD - Interstitial lung disease; HRCT - High-resolution computerized tomography; NSIP - Nonspecific interstitial pneumonia; UIP - Usual interstitial pneumoniae; LIP- Limphocytic interstitial pneumonia; PFT - Pulmonary function test; FVC - Forced vital capacity; DLCO - Diffusing capacity for carbon monoxide; MTX - Methotrexate; RTX - Rituximab; TCZ – Tocilizumab.
At time of diagnosis only 2 patients had arterial pressure of oxygen below 60 mmHg on room air and only 3 patients had minor hypercapnia.
Pulmonary hypertensionNine patients (12%) had signs of pH on transthoracic echocardiography (pulmonary artery systolic pressure ≥40 mmHg), with a mean pressure of 60.8 ± 23.8 mmHg. Five patients had SSc and the other 4 had mixed CTD or overlap syndrome with a predominance of scleroderma findings. Echocardiographic data was missing in 13 patients.
Five patients underwent right heart catheterization (RHC). Two of them did not have a confirmation of PH, the others had a mean pulmonary artery pressure of 40 ± 2.1 mmHg and a mean pulmonary vascular resistance of 10.4 ± 6 Wood Units. Of these 3, all with SSc, 1 was classified as having group I PH, the others were group III. In the remainder 4 patients with echocardiographic signs of PH, RHC was not conducted as the procedure was considered too risky or of no added benefit to the management of these particular patients.
AutoantibodiesAnti-nuclear antibodies were positive in 79% of patients (titre ≥1:320 in 80%).
Amongst SSc patients, 54% were anti-Scl70 positive, 15.4% were anti-centromere positive and 7.7% were anti-PM-Scl positive.
Amongst RA patients, 73.3% were simultaneously rheumatoid factor (mean value of 338 ± 266 UI/mL) and anti-citrullinated protein (anti-CCP) positive (mean value of 170.1 ± 160.4 UI/mL)
In overlap syndromes, the most frequently found serologic markers were anti-SSA (present in 40% of patients) followed by anti-SSB, anti-Scl70, anti-PM-Scl, rheumatoid factor and anti-CCP (all present in 20% of these patients). All patients classified as having mixed CTD were anti-RNP positive. Four out of the five patients with autoimmune myopathy were diagnosed with anti-synthetase syndrome (3 were anti-Jo1 positive and the other was anti-PL12 positive). The patient with polymyositis was anti-Ku positive (anti-MDA5 was not tested).
TreatmentSystemic corticosteroids were by far the most used drug, being used at some point during the course of disease in 86.7% of the patients. Mycophenolate mofetil was used in 36% of patients, cyclophosphamide in 33% and azathioprine in 29.3%. Methotrexate (MTX) was used prior to ILD diagnosis in 32% of patients (n = 23), but was then switched to another immunossupressor drug (mainly mycophenolate) in all but 2 patients. Out of the patients who were treated with MTX, 3 (13%) had lung fibrosis considered to be caused by the drug.
Biologic drugs were used in 39% of the patients, with tocilizumab and rituximab being the most used as first-line therapy (13 patients each), followed by etanercept (3 patients). Sixty percent of the patients with RA were on biologic treatment, as were 23.1% of SSc. Tocilizumab was the most used biologic in RA but this decision was driven by joint disease in all but one patient. All patients with ANCA-positive vasculitis and anti-synthetase syndrome were on rituximab (Table 1). Five patients required a biologic switch due to failure of first-line choice (3 patients were switched from tocilizumab to rituximab, 2 patients were switched from etanercept to tocilizumab). In 3 of these patients the switch was decided based on ILD progression, in the other 2 patients the decision was made due to ongoing activity of the joint involvement (both patients had overlap syndrome). Throughout the follow-up period there were no exacerbations of the ILD caused by the starting of biologic treatment.
Two patients with SSc were recently treated with nintedanib (one with UIP pattern, the other NSIP).
Six patients of the nine diagnosed with pH were prescribed specific treatment targeted at PH: 3 bonsentan, 1 alprostadil, 1 iloprost and 1 sildenafil. The last two patients were later switched to bonsentan.
MortalityThis cohort had a mortality rate of 20% (15/75 patients) during the analyzed period, with a mean survival of 67.8 ± 57.3 months since CTD diagnosis and 37.8 ± 20.9 months since ILD diagnosis. Mortality rate was higher in RA and overlap syndrome (Table 1).
Comparing deceased patients with the survivors, the first group was older at the time of CTD diagnosis (58.6 ± 4.9 vs 51.5 ± 2.7 years-old, p = 0.23), older at the time of last follow-up (66.4 ± 11.1 vs 60.4 ± 15.2 years-old, p = 0.03) and more frequently male (33 vs 20%, p = 0.31). UIP pattern on HRCT was related with higher mortality but the difference was not significant. There were no significant differences regarding other imaging patterns, diagnosis of SSc, corticotherapy and ANA positivity. The presence of pH was clearly associated with higher mortality (OR 14.41, p = 0.006) and so was the use of biologics (OR 5.56, p 0.025).
DiscussionThis population of 75 CTD-ILD patients constitutes a relevant sized population with a long mean follow-up time of 49 ± 31 months over the span of 6 years. ILD was diagnosed prior to CTD in only 8% of patients but this is probably biased since this unit is primarily focused on CTD with a background in internal medicine. More importantly, diagnoses were simultaneous in 35% of patients, emphasizing the relevance of lung involvement in these conditions. It is important to note that all patients had a definite diagnosis of a CTD, hence no patient fell within the recently suggested definition of “interstitial pneumonia with autoimmune features”.20
Most patients presented with a NSIP pattern on HRCT but more common findings such as pleural thickenings and effusions can point towards a possible underlying CTD in an ILD patient. The majority of SSc patients had extensive lung disease on HRCT at the time of ILD diagnosis, again reinforcing the relevance of this condition, particularly in light of the fact that it can be clinically silent during a significant amount of time.3 Lung biopsy was seldom used and rarely provided additional information.
Regarding PFT, an isolated diminished DLCO (35% of patients) may be the only abnormality. ANCA-positive vasculitis and mixed CTD were the subgroups with the most impactful disease at diagnosis. In ANCA-positive vasculitis the severity of the lung disease was mainly on account of a very low DLCO, which was probably caused by vasculitic disease rather than ILD per se.
Regarding treatment, corticosteroids remain the first-line but the use of other immunosuppressants such as mycophenolate mofetil is increasing.
Methotrexate was stopped in most patients upon ILD diagnosis, even if only 3 patients presented with fibrosis admitted being related to it. Over the years MTX-associated lung disease has been a controversial topic, with earlier studies pointing towards a slightly increased risk of lung fibrosis.21 MTX was stopped for most patients to prevent it from being a confounding factor in the follow-up and because safer and equally effective drugs were available. In retrospect, and in light of a recent multivariate analysis showing that there was no association between development of RA-ILD and MTX use and that the drug could in fact have a protective role, the drug could have been safely continued.22
Biologic drugs are being increasingly used in CTD-ILD. The most used ones were tocilizumab and rituximab and their use was mainly dictated by the extra-pulmonary manifestations of the underlying CTD—tocilizumab was predominantly used in RA and rituximab for vasculitis and autoimmune myopathies. In this population, biologics were deemed safe to start in patients with ILD as there were no exacerbations upon the start of biologics, contrary to some prior reports.23 The use of biologics was associated with worse survival but this is probably related to disease severity rather than drug-related complications.
The antifibrotic nintedanib was used in 2 SSc patients with extensive lung fibrosis, after positive safety results in idiopathic pulmonary fibrosis,24 and might be a promising option for slowing ILD progression in these patients, as shown by the SENSCIS Trial.17 However, criteria for nintedanib use in SSc-ILD is up for debate,25 and both our patients remained on background immunossuppressive treatment. Evidence is emerging for the use of antifibrotics in other CTDs. The recently published INBUILD Trial showed that nintedanib leads to a lower annual rate of decline in FVC in patients with progressive fibrosing interstitial lung diseases, including patients with autoimmune diseases, the majority of them with RA-ILD and also including patients with mixed CTD.26
With 20% mortality rate and a mean survival of 37.8 ± 20.9 months, ILD constitutes an important burden on CTD patients and significantly impacts their prognosis. In this cohort, pH was the single most important mortality predictor (OR 14.41, p = 0.006).
Even though this article provides an overview of the general population encountered at an autoimmune diseases unit, some particular subsets of patients are not as strongly represented in the cohort (e.g. autoimmune myopathies) and this could lead to an underrepresentation of more specific and less common findings, as is the case of organizing pneumonia. At the other end, it could lead to an overrepresentation of rare findings, such as the LIP pattern in a patient with polymyositis. Heterogeneity regarding the underlying CTDs, disease duration, treatment and follow-up and the retrospective nature of the study may limit the interpretation of some of the results. The data presented reflects the clinical expertise of a single institution that has its main focus on CTD and may not be fully representative of practices elsewhere.
ConclusionTo the best of our knowledge this is the largest cohort of CTD-ILD presented by a Portuguese centre.
ILD is a potential complication of virtually every CTD, constitutes an important burden on these patients and significantly impacts their prognosis. A systemic and multidisciplinary overview is essential for adequate management, in order to fill in the remaining gaps regarding early diagnosis, follow-up and treatment of these patients.
Author contributionsRPO conceived the idea for the manuscript, collected the data, wrote the first draft and co-wrote the paper. RR collected the data and co-wrote the paper. LM collected the data, was responsible for statistical analysis and co-wrote the paper. BG, SO and JDA conceived the idea for the manuscript. All authors reviewed and approved the final version of this manuscript.
Conflicts of interestsThe authors have no conflicts of interests to declare.
The authors would like to thank the Department of Respiratory Medicine of Hospital Prof. Doutor Fernando Fonseca for the collaboration throughout the years.


