



Case report of a male patient with a five-decade follow-up history in a tertiary care hospital distinguished for malabsorption syndrome, failure-to-thrive, meningitis and recurrent bacterial, fungal and mycobacterial pulmonary infections. Additionally, he developed epidermodysplasia verruciformis, several in situ spinocellular carcinomas and an uncharacteristic parenchymal lung disease. Surgical lung biopsy suggested pulmonary alveolar proteinosis with fibrotic change. Retrospectively, severe monocytopenia had been overlooked in the past, as well as low B and NK cell blood counts. Flow cytometry confirmed the absence of the previous cell subsets along with an undetectable population of dendritic blood cells.
Dendritic cell, monocyte, B and NK lymphoid Human Deficiency Syndrome (DCMLS) is a novel rare immunodeficiency described in 2010, linked to GATA-2 mutation. This syndrome should be highlighted as a rare cause of acquired PAP, with a radiological pattern encompassing potential fibrotic change. Failure to recognize monocytopenia may impede the chance to diagnose.
Relato de um caso clínico de um doente do sexo masculino com um historial de acompanhamento de cinco décadas, num hospital de cuidados terciários, caracterizado por um síndrome de malabsorção, insuficiência de crescimento, meningite e infecções pulmonares bacterianas, fúngicas e micobacterianas recorrentes. Além disso, desenvolveu epidermodisplasia verruciforme, diversos carcinomas espinocelulares in situ e uma doença pulmonar parenquimatosa não definida. Uma biópsia pulmonar cirúrgica sugeriu uma Proteinose Alveolar Pulmonar com alterações fibróticas. Em retrospectiva, uma monocitopenia grave negligenciada no passado, bem como uma baixa contagem de células B (linfócitos B) e NK (células ‘natural killer’). Uma citometria de fluxo confirmou a ausência dos subconjuntos de células anteriores, juntamente com uma população indetectável de células dendríticas sanguíneas.
A Síndrome de Deficiência Humana de células dendríticas, monócitos, linfóides B e NK é uma rara imunodeficiência descrita recentemente em 2010 e relacionada com a mutação do gene GATA-2. Esta síndrome deverá ser referida como uma causa rara de PAP (proteinose alveolar pulmonar) adquirida, com um padrão radiológico que poderá apresentar uma potencial alteração fibriótica. A ausência de reconhecimento da monocitopenia pode impedir a oportunidade de chegar a este diagnóstico.
We present the case report of a 61-year-old male patient with a five-decade history of medical follow-up in a tertiary care University Hospital who is a non-smoker without relevant family history or occupational exposures. The history of the present illness started at the age of 9, with a diagnosis of pulmonary tuberculosis (PT) for which standard antibacillary treatment was completed. He presented failure to thrive in his early childhood, and at the age of 18, he was admitted twice in the Internal Medicine Department for diarrhea, malabsorption syndrome and acute tracheobronchitis. By the age of 20 he had pneumococcal meningitis, but had recovered without neurological sequelae. During the following decade he had further admissions for community-acquired pneumonias, “middle lobe syndrome” and otitis media.
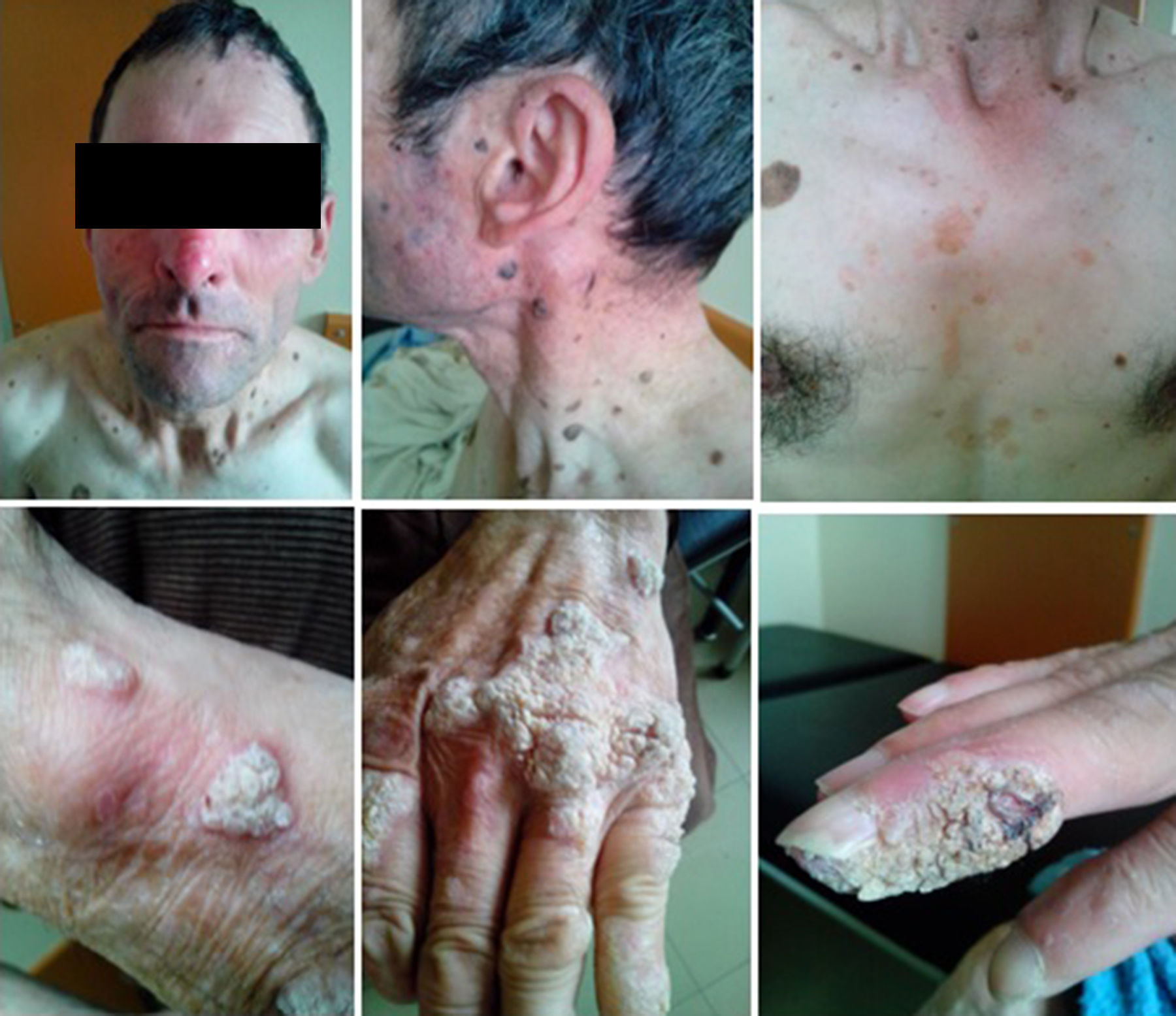
At the age of 37 he was again diagnosed with PT and completed standard treatment, supervised by the Tuberculosis District Center. In his forties he started presenting recurrent processes of bronchitis with isolation of Haemophilus influenzae and Streptococcus pneumoniae. He also started dermatological consultations for epidermodysplasia verruciformis that gradually developed on his hands, upper limbs, torso, and neck, and severe perineal condylomas that were repeatedly treated with electrodessication and curettage (Fig. 1). Since then, he has been diagnosed six times with in situ cutaneous spinocellular carcinoma. His skin biopsies typically revealed dyskeratotic spinocellular carcinomas and “bowenoid papulosis” of a verruciform nature, with abnormalities in the granulosa cell layer suggesting infection by human papillomavirus (HPV).
At the age of 47, one of his admissions to the Pulmonology Department was for Aspergillus fumigatus lung infection (the patient did not fulfill the criteria for allergic bronchopulmonary mycosis). A complete investigation for cystic fibrosis was performed and, after a borderline sweat test, he showed normal spermogram and negative Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) genotype mutation study.
Gradually, he started developing parenchymal lung disease, initially with reticulomicronodular pattern with upper lobe predominance on chest radiograph. In his fifties, computed tomography (CT) scan showed a septal thickening pattern with reticulation, traction bronchiectasis and micronodulation. Pulmonary function tests presented moderately severe restriction by spirometry (Tiffeneau index of 0.74, Forced Vital Capacity 57.2%, forced expiratory volume in one second 52.8%), corrected to normal by plethysmographic lung volumes (total lung capacity 80.8%, residual volume 109.2%), with a moderately affected diffusion capacity (DLCO 54%), while maintaining satisfactory blood gas analysis at room air (pO2 80mmHg, pCO2 41mmHg, SaO2 96%).
At his last admission, at the age of 61 years, he presented a more pronounced diffuse interstitial reticulomicronodular pattern and progressive emaciation. He complained of productive cough with high volume purulent sputum and worsened dyspnea (grade III mMRC). High-resolution chest CT showed a more profuse septal thickening, micronodular pattern of random distribution, “tree-in-bud” images, along with apical subpleural blebs, linear fibrotic lines and distortion of the normal bronchovascular architecture (Fig. 2). Flexible bronchoscopy presented inflammatory changes of the mucosa and mucopurulent sputum. BAL, in the context of infection, showed a concordant neutrophilic cellular profile (860,000total cells/mL, with 84% neutrophils). The cytopathological study was negative and microbiological workup allowed for the identification of Corynebacterium species and Acinetobacter baumannii. Serum immunoglobulins were normal, as were complement levels and serum angiotensin-converting enzyme titers. There were positive anti-nuclear antibodies with positive anti-cytoskeleton fibers and anti-vimentin antibodies, and negative anti-neutrophil cytoplasmic antibodies (ANCAs). His serological panel was negative for syphilis, B and C hepatitis and Human Immunodeficiency virus (HIV)-1/2 infection, but presented high IgM and IgG titers for cytomegalovirus, Epstein–Barr and herpes simplex virus-1.
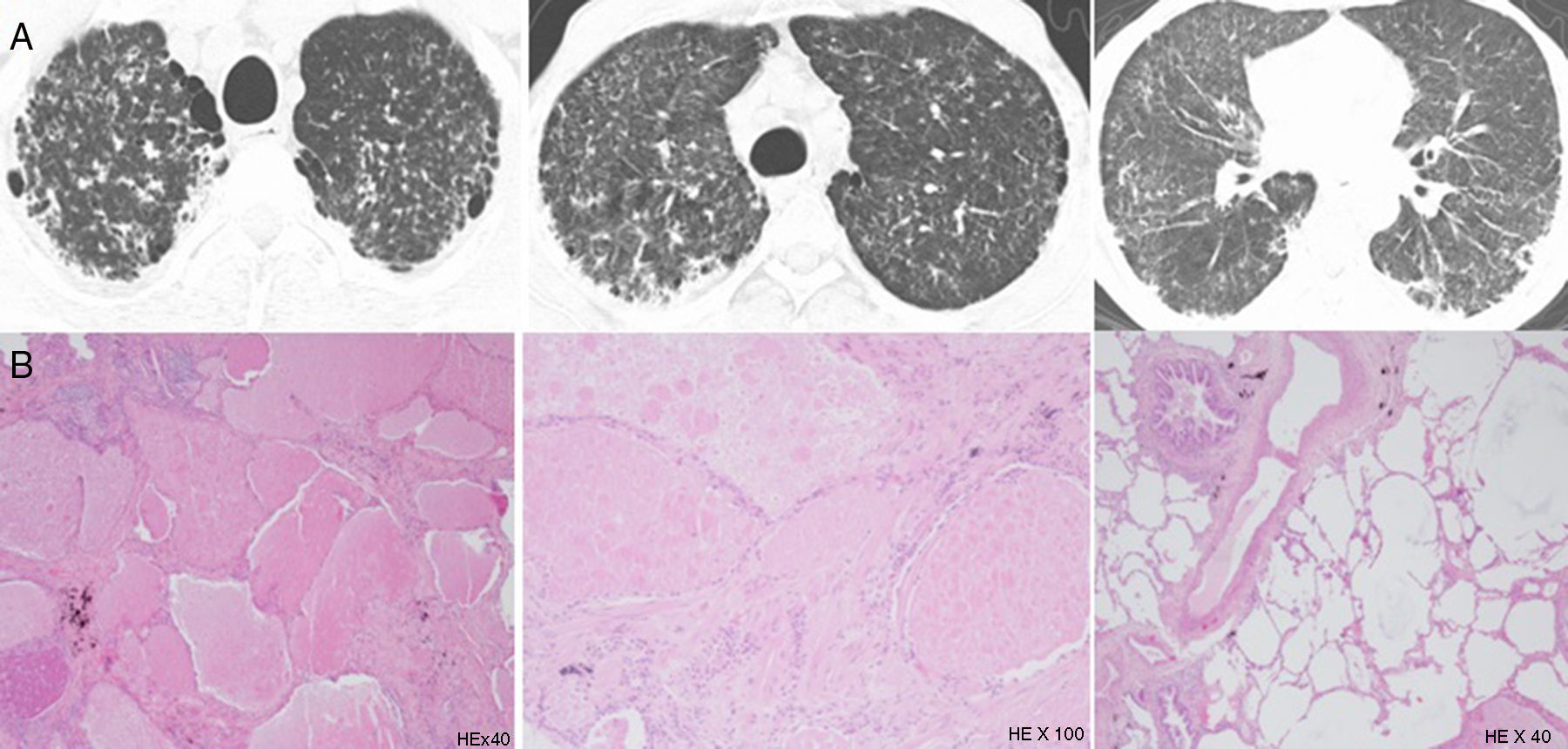
Patient's parenchymal lung disease – imaging and lung biopsy. (A) Chest CT images with interlobular thickening pattern with reticulation, micronodulation and small scattered areas of ground-glass and lobular opacities. Alveolar proteinosis depicted by diffuse alveolar flooding by eosinophilic proteinaceous material, chronic lymphocytic infiltrate and septal fibrosis with collagen deposition.
He underwent a multiple-lobe surgical lung biopsy, which was suggestive of alveolar proteinosis with some fibrotic trace. It revealed aspects of diffuse alveolar occupation by eosinophilic, Periodic-Acid-Schiff positive proteinaceous material, with a light macrophagic reaction, together with abnormal lobular architecture, diffuse septal fibrosis with focal collagen deposition and some multinucleate foreign body giant cells with cholesterol crystal clefts (Fig. 2). There were no granulomas.
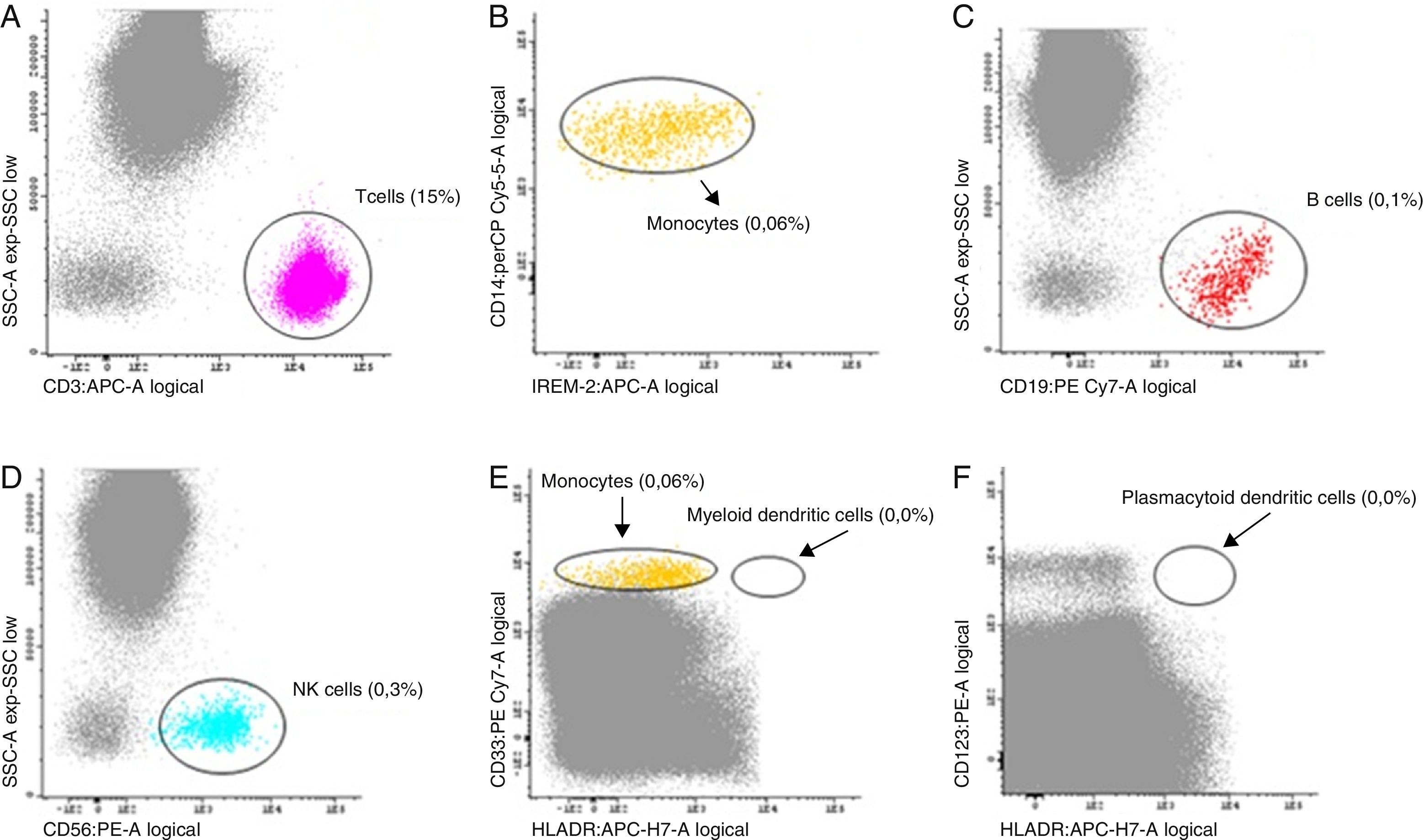
By this time absent serum monocytes and low B and NK cell counts had been acknowledged. Indeed, severe monocytopenia had been ignored for years, as had low B and NK cell blood counts on previous blood workup. At this moment, suspicion about a possible immunodeficiency syndrome associated with diffuse parenchymal lung disease was assessed. A peripheral blood sample was studied by a detailed flow cytometry at a Histocompatibility Center Laboratory (Fig. 3). It revealed an almost complete absence of monocytes (CD14+, CD300E+), B cells (CD19+CD20+) and NK cells (CD56+, CD3−). Additionally, there was no detectable myeloid or plasmacytoid population of dendritic cells – DCs – (CD16+).
Bone marrow biopsy disclosed hypocellularity with megakaryocytic dysplasia and maturative abnormalities. Marrow immunophenotyping revealed very scarce monocytes (0.4%), mainly consisting of immature forms (promonocytes), absence of CD34+ B cell precursors, and scarce myeloid DCs and absent plasmacytoid DCs or CD34+ cells compromised with this lineage.
The patient fulfilled the diagnostic criteria for Dendritic cell, Monocyte, B and NK Lymphoid Human Deficiency (DCML) Syndrome, as suggested by Bigley and Collin.1,2 He presented cutaneous epidermodysplasia with several in situ carcinomas; mycobacterial infection – two episodes of PT; severe blood monocytopenia, B cell and NK cell deficiencies; absolute blood DCs deficiency; hypocelullar bone marrow with megakaryocytic dysplasia; and normal immunoglobulins.
DCML, also known as “autosomic dominant and sporadic monocytopenia”3 or “MonoMAC”4, is a novel immunodeficiency syndrome, primarily described in 2010 by Vinh and colleagues.3 It results from one of several known GATA-2 mutations5 determining a progressive and selective loss of bone-marrow multi-lymphoid progenitors – pluripotent cells that originate DC, monocytes and lymphoid cells – and partial depletion of granulocyte-macrophage progenitors (although sufficient to sustain granulopoiesis).2
The associated deficit of DCs and the understanding of the immunoregulatory mechanisms were highlighted in 2011 by Bigley.1 DC depletion is associated with a marked increase in fms-like tyrosine-kinase ligand (FLT3L) and concomitant loss of regulatory T cells, which can contribute to auto-immune phenomena.2,6,7 Remarkably, despite the near absence of monocytes, tecidual macrophages – like alveolar macrophages – are preserved but are probably functionally defective, explaining the possible feature of PAP.2 It has been suggested that tecidual macrophages may originate from local proliferation.
This progressive form of bone marrow lymphoid failure, at a given point in time, assumes features of a distinct form of myelodysplasia4 with composed deficiency of monocytes, B, NK and DCs subsets, along with proneness to mycobacterial, fungal and viral infections, epidermodysplasia verruciformis and in situ carcinomas related to chronic HPV infection, and the possible development of PAP in a significant number of patients. Cases may be sporadic or of autosomal dominant heredity, and tend to present in the third or fourth decades of life – although previous unnoticed monocytopenia might have been present several decades earlier along with an ill-defined history of unusual infections starting at an early age.3
Unlike monocytopenia, NK and B cell deficiencies are generally not easily detected in regular blood counts due to the preservation of peripheral T cells. Patients tend to present normocellular or hypocellular (89%) marrows, always with megakaryocyte dysplasia.4
Severe perineal HPV infection from adolescence is a common feature, progressing with variously diffuse epidermodysplasia verruciformis, apparently as a result of the combined monocyte and NK cell deficiencies.8 Weight loss and constitutional symptoms are frequently reported,3 but the natural history is marked by prominent recurrent infections by mycobacteria, aerobic bacteria and fungus. Although not an universal finding, mycobacterial infection is seen in the majority of patients1 and possibly derives from defective phagocytosis resulting from GATA2 mutation,9 as well as from impairment of the IL-12/INF-γ axis induced by the cellular deficiencies.2
In advanced disease, progression to acute myeloblastic leukemia is frequent and autoimmune phenomena – lupus-like syndrome, multiple sclerosis – may also occur in a fraction of patients.3
Regarding the emergence of parenchymal lung disease in DCML, PAP occurs in 33% of patients (36% in sporadic and 29% in autosomal-dominant patients), with a median onset age of 42 years, and without detectable anti granulocyte-macrophage colony-stimulating factor (GM-CSF) antibodies.3 The impossibility to perform anti-GM-CSF antibody testing at our Center is a limitation in this case report. However, as the patient fulfilled all the diagnostic criteria proposed by Bigley and Collin, we can speculate that anti-GM-CSF antibodies would probably not be present in this case either, according to what is reported on patients with DCML.
It apparently results from monocyte/macrophage dysfunction3 with metabolism and phagocytic impairment (induced by GATA-2 mutations),9 although some believe that mycobacterial infection may play a pathogenetic role. Therapeutic administration of GM-CSF does not seem to have a significant effect, but whole-lung lavage may present moderate success.3
A remarkable aspect of this case is that the patient's diffuse parenchymal lung disease was histologically consistent with chronic PAP that evolved to fibrosis. As already suggested by some authors, established fibrotic burden probably results from the tecidual reaction to long-lasting subclinical proteinosis, although it is possible that the history of recurrent bacterial and mycobacterial infections may have also contributed.10 Generally, PAP CT pattern is not strictly alveolar as many patients present a “mosaic attenuation pattern” with ground glass opacification, or airspace consolidation, over a background of thickened interlobular septae.11,12 Moreover, there are various reports of pulmonary fibrosis of various degrees associated with PAP, sometimes diagnosed several years after disease onset.10–17 In some cases the onset of PAP was clearly established prior to fibrosis, using serial lung biopsies.15,17 A review by Holbert regarding CT findings in a small group of patients with PAP found substantial fibrosis in 2 cases.10 Interestingly, a recent paper from Ishii also found that a marked cystic and interstitial appearance might be particularly related to hereditary or secondary forms of PAP.18 Thus, a lung fibrotic pattern may be encountered in the context of chronic secondary PAP (sometimes subclinical), namely in the context of DMCL syndrome.
Regarding treatment options, hematopoietic stem cell transplantation is seen as potentially curative for DMCL patients, even for subjects with established respiratory failure.1,3
This entity should be considered as a possible etiology of unexplained persistent monocytopenia, high susceptibility to opportunistic infections and simultaneous epidermodysplasia verruciformis, with possible development of secondary PAP potentially progressing to fibrotic change. Further characterization of DMCL syndrome, studies addressing the pathogenetic mechanisms of interstitial fibrosis in patients with PAP and the different reactions of lung parenchyma in this disease will be important in the near future.
At the time of this publication, arrangements are being made to investigate the specific GATA-2 mutation involved in this patient. Ideally, genetic testing should always be accomplished in order to expand the knowledge of orphan diseases such as DCML.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.


