

Uma mulher de 68 anos foi submetida a uma ressecção de um linfoangioendotelioma mediastinal observado na monitorização de uma lobectomia inferior esquerda devido a bronquiectasia, complicada por quilotórax. Isto levou a uma reavaliação do espécime pulmonar que revelou, além da bronquiectasia inflamatória, nódulos de pequenas células fusiformes no parênquima pulmonar, semelhantes a nódulos pulmonares de tipo meningotelial, mas com positividade imunohistoquímica para actina do músculo liso. A hipótese de desenvolvimento inicial de linfangioleiomiomatose pulmonar é discutida.
A 68 year old woman was submitted to a mediastinal lymphangioleiomyoma resection found in a follow-up study of lower left lung resection due to bronchiectasis complicated by chylothorax. This led to a revaluation of the pulmonary specimen that revealed, in addition to inflammatory bronchiectasis, small spindle cell nodules in the lung parenchyma, similar to minute pulmonary meningothelial-like nodules, but with smooth muscle actin immunohistochemical positivity. The possibility of initial pulmonary development of lymphangioleiomyomatosis is discussed.
A linfangioleiomiomatose (LAM) é uma doença rara de etiologia desconhecida, que incide, classicamente, em mulheres na idade reprodutiva e, ocasionalmente, nas pós-menopáusicas, com uma incidência estimada de 1-2,6 /1.000.000 mulheres. Os 2 formas de apresentação mais comuns são a dispneia de esforço e o pneumotórax. Outros sintomas menos comuns incluem hemoptise, tosse não produtiva, quilotórax e ascite quilosa1. As alterações microscópicas consistem na proliferação de células musculares lisas imaturas na parede das vias aéreas, vénulas e linfáticos do pulmão, originando diminuição das vias aéreas, obstrução e aprisionamento do ar e, num estádio mais tardio, lesões pulmonares quísticas e quistos contendo flúido linfático. A imagiologia reconhece o padrão, na maior parte das vezes, com alterações atingindo com maior frequência os lobos inferiores. A doença progride insidiosamente até à insuficiência respiratória, sendo o estádio terminal atingido num período que varia de alguns anos a cerca de 3 décadas1.
A LAM pertence ao grupo dos PEComas, caracterizados pela presença de 2 tipos celulares: células epitelióides, tipicamente de localização perivascular, e células fusiformes, semelhantes a células do músculo liso, envolvendo o primeiro tipo celular. Há grandes variações no volume dos 2 tipos celulares em cada tumor. Estas células são tipicamente positivas para marcadores melanocíticos (HMB-45, Melan-A, MITF e NKI/C3) e marcadores musculares (α-SMA e calponina), sendo menos frequente a expressão de desmina. A proteína S100 e as citoqueratinas estão normalmente ausentes. A nível ultraestrutural as células têm glicogénio citoplasmático abundante, pré-melanossomas, filamentos finos com corpos densos ocasionais, hemidesmossomas e junções intercelulares fracas. Os critérios de malignidade listados pela OMS para os PEComas resultam da combinação dos seguintes fatores: crescimento infiltrativo, hipercelularidade marcada, tamanho nuclear aumentado/hipercromasia, atividade mitótica elevada, figuras mitóticas atípicas e necrose de coagulação2.
A LAM e o complexo da tuberose esclerosa (TSC) poderão partilhar uma relação genética comum; a TSC é causada por mutações germinativas nos genes supressores tumorais TSC1 e TSC2, localizados nos cromossomas 9q34 e 16p13, respetivamente, e os tumores ocorrem por perda de heterozigosia em um dos genes. O gene TSC2 foi implicado na etiologia da LAM, uma vez que mutação e perda de heterozigosia foram demonstradas nas células em proliferação. A patogénese exata da LAM não está ainda definida, mas informação acumulada confirma o papel da mutação somática do gene TSC2, sugerindo uma mutação espontânea numa célula pulmonar previamente normal, predispondo à disfunção da supressão tumoral e proliferação local anormal1,3; foi descoberto que quando há uma deficiência nas proteínas hamartina-tuberina, uma ativação anómala do mTOR leva ao crescimento celular descontrolado4. A expressão variável dos fatores de crescimento e seus recetores na LAM pode resultar em funções específicas que facilitam as ações fibrogénicas, proliferativas e de regulação da matriz pelas células tumorais1.
Uma lesão que também contém células epitelióides5 ou células fusiformes com núcleo oval a indentado e alongado6 são os «minute pulmonary meningothelial-like nodules» (MPMN); estes formam ninhos ou espirais com disposição de tipo «Zellballen», centradas em pequenas veias, com pressuposta função de monitorização do oxigénio como quimiorrecetores, tendo sido reportadas como «quemodectomas pulmonares»5. No entanto, não têm grânulos endócrinos e não estão associados aos nervos à avaliação por microscopia eletrónica. Estudos ultraestruturais revelam que estas células são semelhantes às meningoteliais. Têm imunorreatividade forte para o antigénio epitelial de membrana e para vimentina, e são negativos para citoqueratinas, proteína S100, NSE e α-SMA. Podem conter uma pequena quantidade de hemossiderina, demonstrável por colorações para o ferro, que evocam uma habilidade fagocítica7.
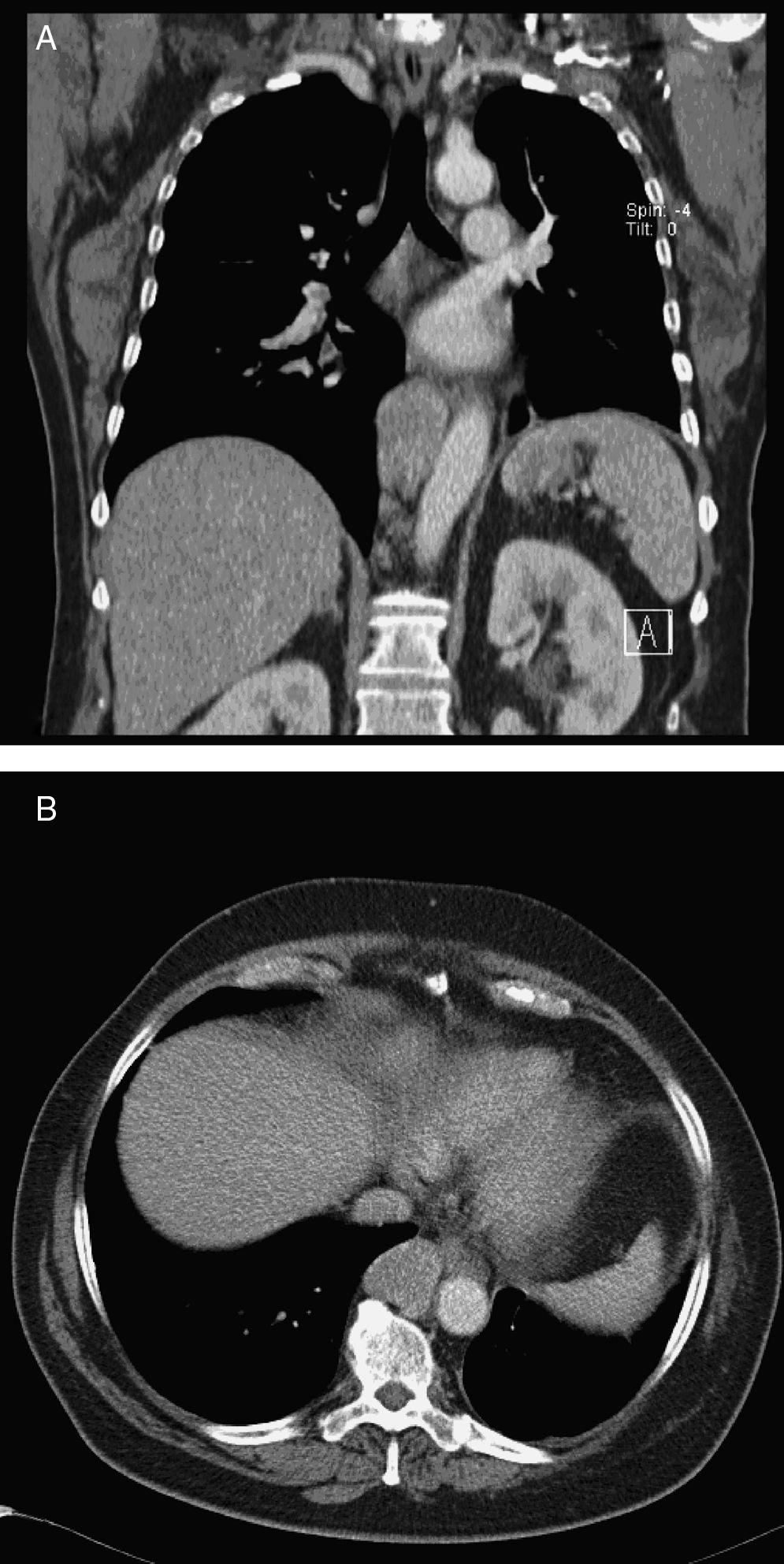
Caso clínicoUma mulher de 68 anos de idade, com história prévia de lobectomia inferior esquerda por bronquiectasia complicada por quilotórax (1994), foi submetida a uma TC de rotina (2008) que revelou uma massa no mediastino posterior inferior direito, com 5cm de diâmetro (fig. 1), que foi submetida a ressecção cirúrgica.
 e axial (B). Linfonodos mediastínicos posteriores alargados (recesso ázigo-esofágico distal).")
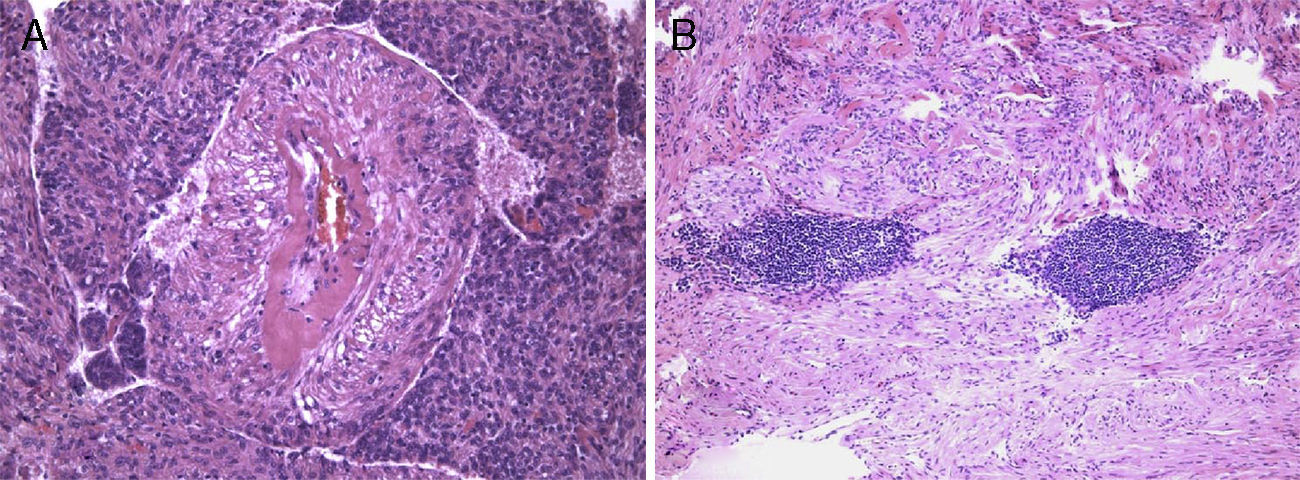
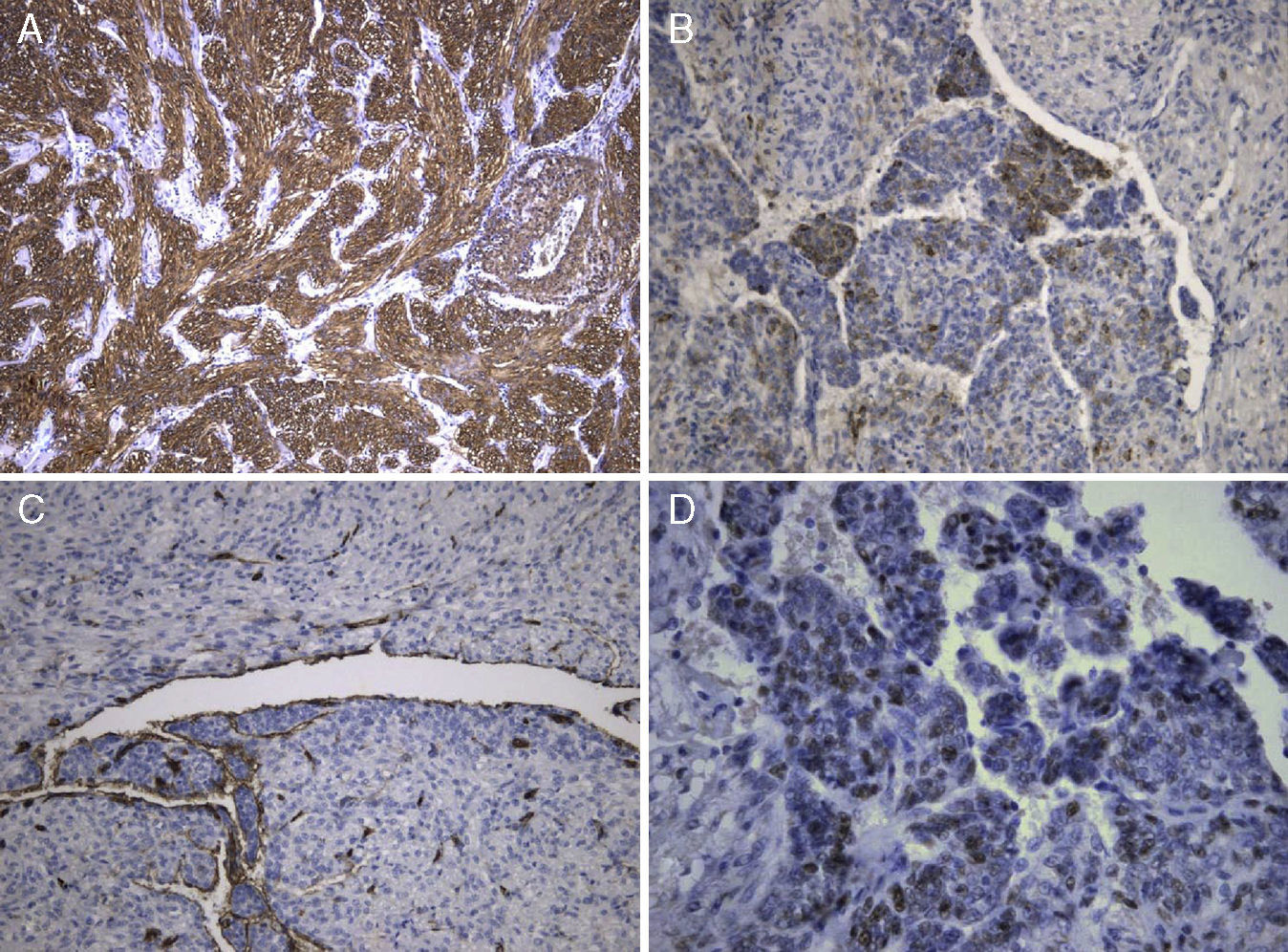
A lesão mostrava tecido rosado e elástico, com áreas friáveis e congestivas focais, ao estudo microscópico sendo constituída por células fusiformes com núcleos ovais e monótonos, que predominavam sobre células epitelióides de núcleos redondos e hipercromáticos; ambos os tipos celulares não exibiam mitoses ou atipia. A vascularização da lesão consistia em vasos de parede colagenizada de lúmen excêntrico, de onde radiavam as células fusiformes, com espaços em fenda presentes por toda a lesão (fig. 2A). Observaram-se raros agregados linfóides centrais e periféricos, caracterizando o tecido linfóide residual do gânglio linfático críptico, onde o processo cresceu de forma expansiva e bem delimitada (fig. 2B); o PAS-diastase revelou a presença de glicogénio em ambos os tipos celulares, e o estudo imunohistoquímico mostrou: positividade forte para HHF-35, desmina e α-SMA (fig. 3A); positividade multifocal para HMB-45 nas células epitelióides (fig. 2B); marcação para CD34 no revestimento interno dos espaços em fenda (fig. 2C); positividade para recetores de estrogénios (fig. 2D) e progesterona, cromogranina e NSE. A marcação para citoqueratinas de baixo peso molecular, CD117 e proteína S100 foi negativa.
 Células fusiformes irradiando da parede espessada de vasos e células epitelióides na periferia. Sem mitoses ou atipia detetadas. B) Vestígios de linfonodo.")
 Positividade citoplasmática para actina de músculo liso. B) Positividade multifocal para HMB-45. A positividade é mais evidente nos agregados epitelióides. C) Fendas com positividade no revestimento para CD34 – Espaços sinusoidais. D) Positividade nuclear para estrogénio.")
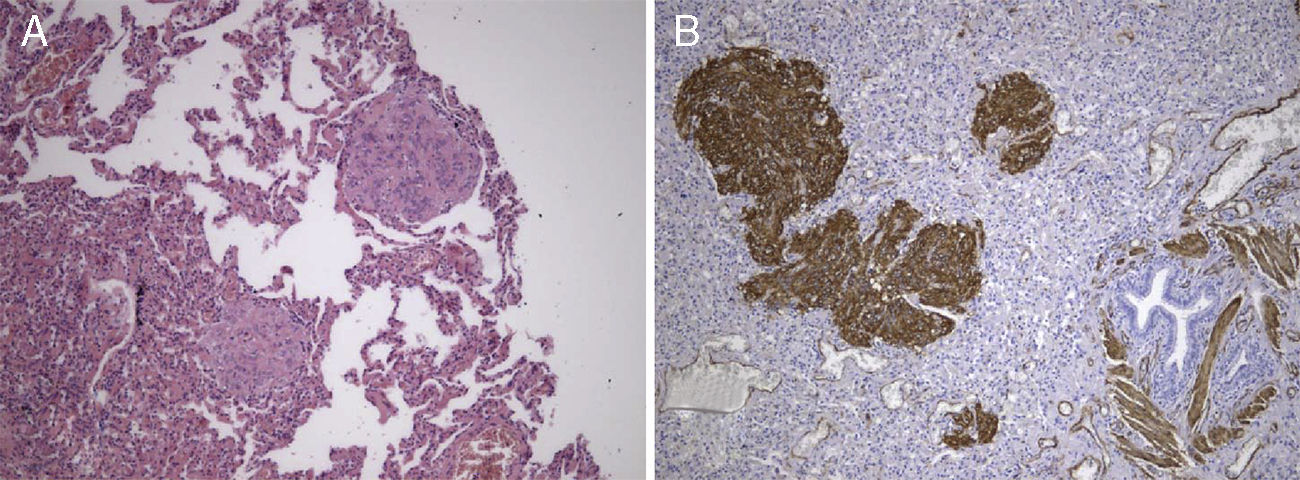
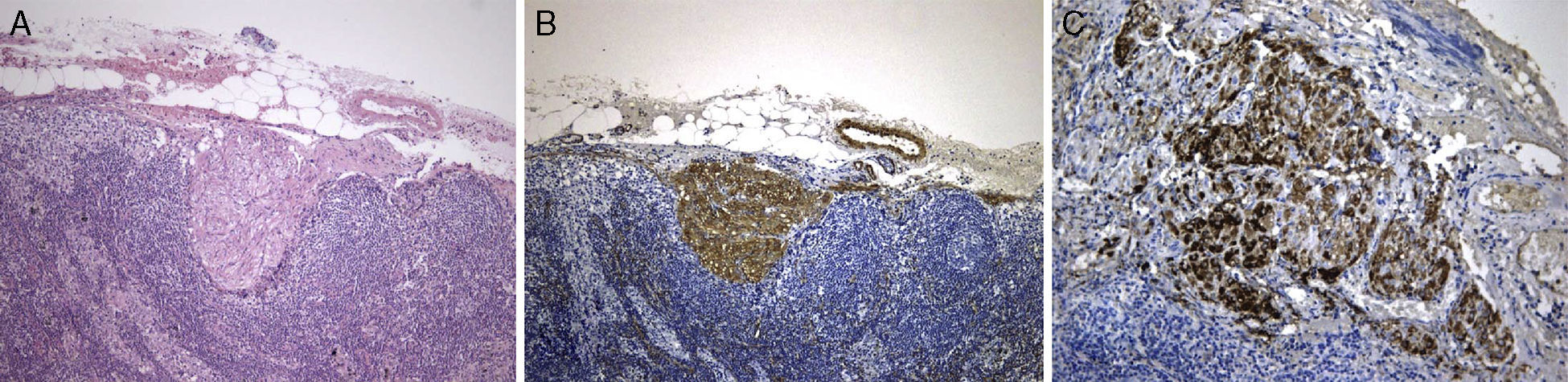
Revimos a peça de lobectomia inferior esquerda, efetuada 14 anos antes, que revelou bronquiectasias inflamatórias, com metaplasia epidermóide focal e um infiltrado inflamatório polimorfo peribrônquico intenso, acompanhado de hiperplasia do tecido linfóide associado ao brônquio (BALT) e abcessos extensos contínuos com o lúmen brônquico. Entre as áreas inflamatórias encontravam-se lesões nodulares espessando os septos alveolares (fig. 4A), consistindo em espirais de células fusiformes, semelhantes às observadas na massa mediastínica. A abordagem imunohistoquímica mostrou positividade para a α-SMA (fig. 4B), marcação duvidosa para HMB-45 e negatividade para a proteína S100. Um gânglio linfático hilar mostrou ainda pequenas áreas de proliferação muscular subcapsular (fig. 5A; B), com marcação positiva para HMB-45 (fig. 5C).
 Proliferação nodular intersticial de células fusiformes. B) Positividade para actina do músculo liso nas células fusiformes nodulares.")
 Proliferação de células fusiformes em linfonodo hilar. B) Positividade para actina de músculo liso. C) Positividade para HMB-45.")
A LAM é considerada uma doença feminina, encontrada frequentemente em mulheres na média dos 40 anos de idade8. Estão reportados apenas 4 casos em homens1. Manifesta-se tipicamente por dispneia progressiva ou pneumotórax recorrente, quilotórax e hemoptises8. Quando envolve gânglios linfáticos ou o ducto torácico, leva à formação de massas quísticas que, quando causam obstrução, originam quilotórax ou ascite1. O diagnóstico definitivo, determinado pelas diretrizes da ERS, requer uma TC de alta resolução (HRTC) pulmonar característica ou compatível e uma biopsia pulmonar com critérios de LAM; ou uma HRTC característica associada a angiomiolipoma renal, efusão quilosa abdominal, linfangioleiomioma/gânglio linfático envolvido por LAM, ou TSC comprovada/provável9. Nos gânglios linfáticos a LAM caracteriza-se por proliferação trabecular de músculo liso, separada por espaços delineados por endotélio sinusoidal10, ocasionalmente com focos de linfócitos dispersos pelas células musculares, um traço representativo do gânglio linfático pré-existente8. No envolvimento pulmonar, a proliferação de músculo liso ocorre nos espaços perivasculares, peribrônquicos e intersticiais, formando nódulos microscópicos nas paredes das vias aéreas periféricas adjacentes aos linfáticos intersticiais2,10. Esta está frequentemente associada a um quadro quístico «enfisematoso». As células fusiformes são regulares e semelhantes a músculo liso, mas podem tomar a aparência de células claras, vacuolizadas ou ocas10. Observa-se expressão imunohistoquímica para marcadores melanocíticos e musculares, recetores de estrogénios e raramente de progesterona2.
Os estrogénios foram considerados como um importante fator na progressão da doença, uma vez que a LAM nunca foi reportada antes da menarca, é sabido acelerar durante a gravidez, e a remissão foi confirmada, em alguns casos, após ooforectomia. Para além disso, recetores de estrogénios e progesterona foram identificados numa subpopulação de células musculares lisas, mesmo quando a doença afeta doentes do sexo masculino1.
Não há tratamento definitivo; apesar de não existir evidência válida da sua eficácia, a progesterona é o tratamento mais frequentemente usado nas pacientes com LAM; os inibidores mTOR, como Sirolimus, deverão ser considerados caso a caso9. As complicações são tratadas sintomaticamente, com o transplante pulmonar oferecendo a única esperança para a cura, com uma sobrevivência de 69 e 58% após um e 2 anos, respetivamente1.
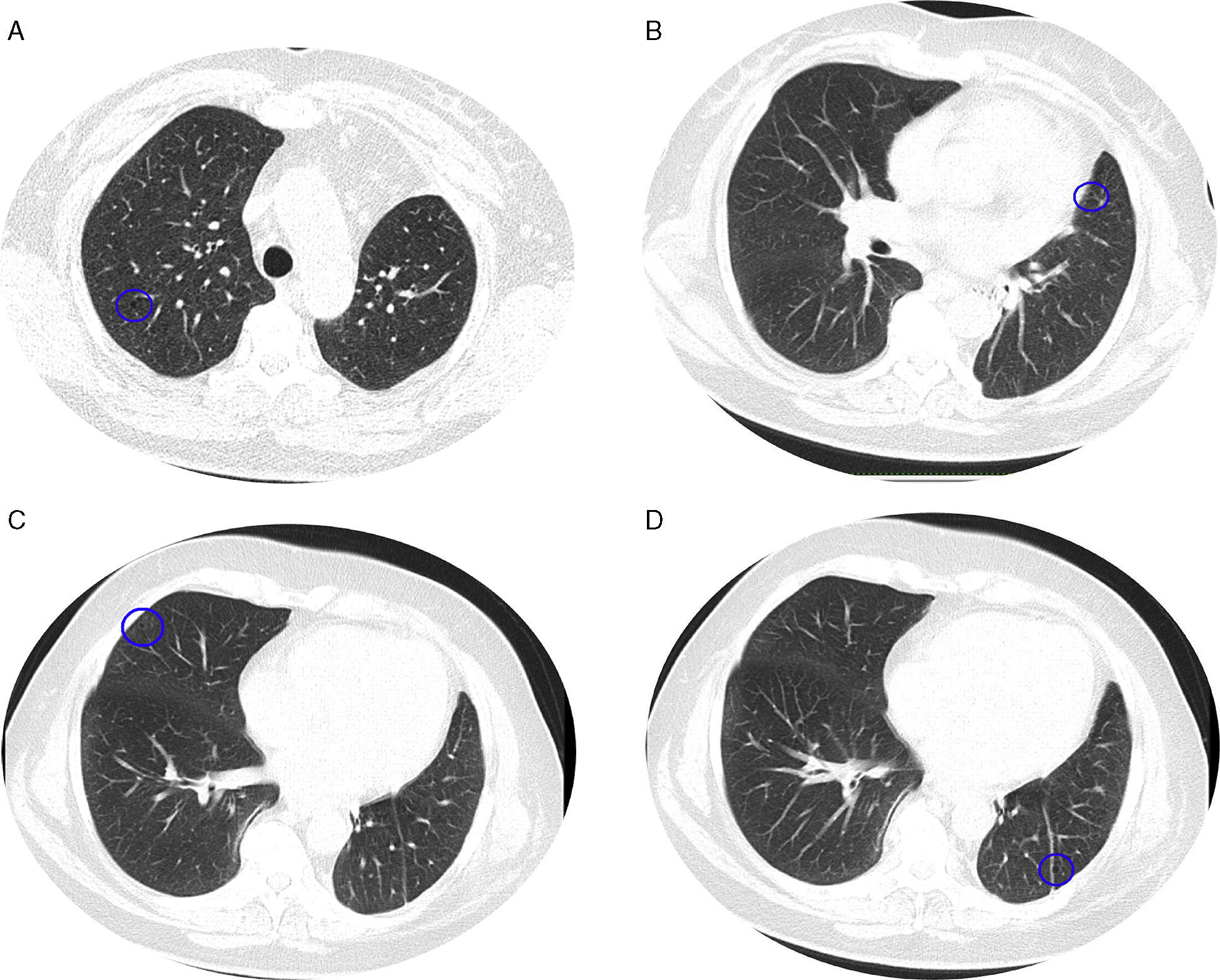
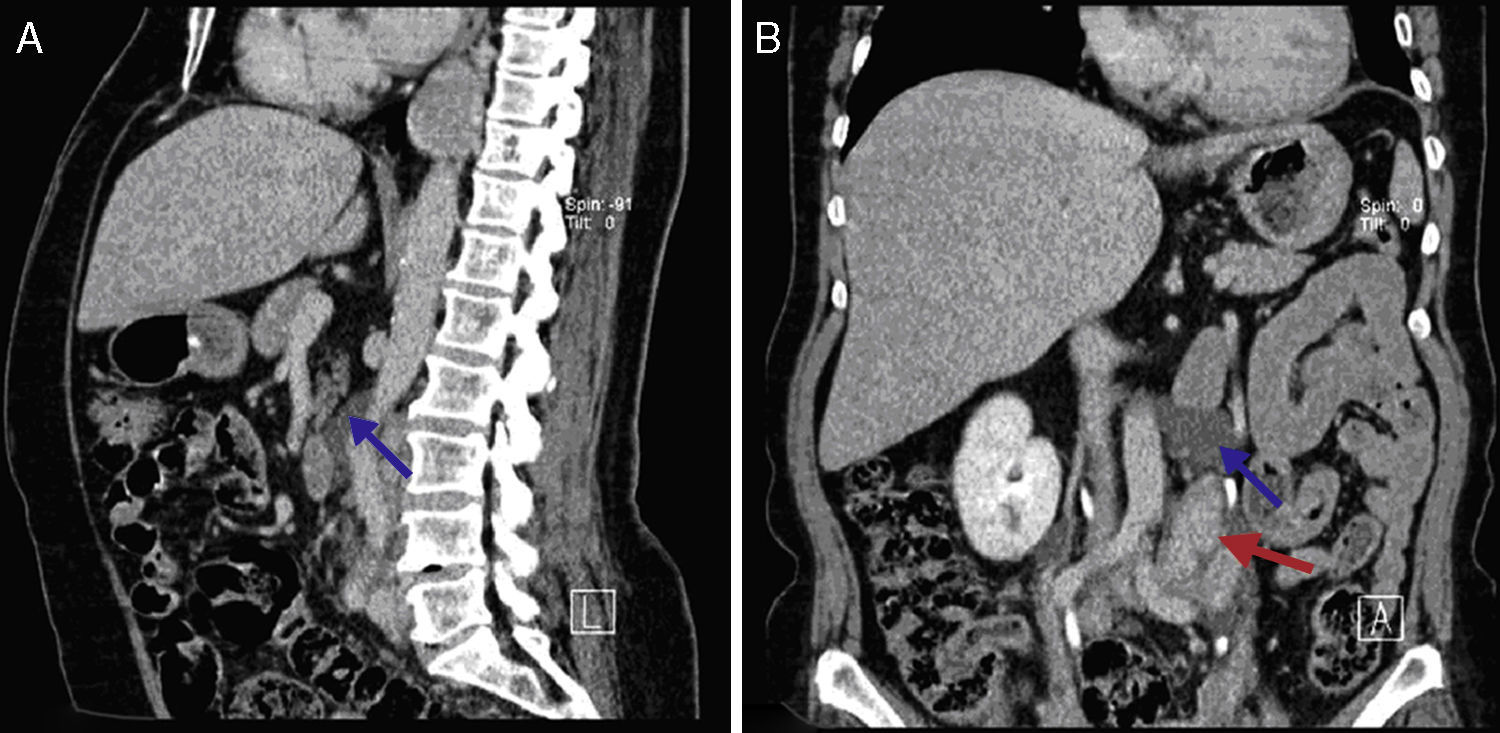
Perante as características morfológicas e imunohistoquímicas, o diagnóstico efetuado foi de um linfangioleiomioma ganglionar. Considerando que o envolvimento extrapulmonar exclusivo é raro5, procedemos à revisão da lobectomia, realizada 14 anos antes por bronquiectasias complicadas de quilotórax. Foi também observada uma proliferação pulmonar nodular intersticial de células fusiformes, inicialmente interpretadas como MPMNs, secundários a hipoxémia local, mas revelando diferenciação muscular com positividade para α-SMA, que não se encontra reportada nesta entidade7. Um dos gânglios linfáticos hilares também mostrava pequenas áreas musculares lisas subcapsulares com imunomarcação para HMB-45. A paciente não exibia o aspeto radiológico típico da LAM na primeira cirurgia, mostrando apenas alterações compatíveis com bronquiectasias e derrame pleural. Em 2008, apesar de ter desenvolvido dispneia de esforço, não foram reportadas alterações consistentes com envolvimento pulmonar por LAM (fig. 6). No entanto, avaliando as imagens retrospetivamente e perante um diagnóstico patológico estabelecido de linfangioleiomioma, poderão ser valorizadas de forma diferente raras lucências visíveis, algumas envolvidas por opacidades em «vidro despolido» ligeiras. Estas assemelhavam-se a quistos pulmonares (com uma parede discernível, que é contrária à hipótese de apenas se tratar de destruição pulmonar enfisematosa). Estes eram bem definidos, com pequenas dimensões, similares uns aos outros e dispersos pelos pulmões. Estas alterações são muito ligeiras, não permitindo um diagnóstico imagiológico inequívoco da doença, mas que também não devem ser ignoradas ou desconsideradas, e podem corresponder, de facto, ao comprometimento pulmonar inicial e ligeiro pela LAM. A TC abdominal mostrou também massas consistentes com dilatação de vasos linfáticos abdominais e com gânglios linfáticos aumentados (fig. 7).
 e imagens TAC axiais de alta resolução (A, C): algumas lucências visíveis (círculos azuis) de pequenas dimensões, semelhantes umas às outras e distribuídas ao longo dos pulmões, bem definidas com uma parede discernível, algumas delas rodeadas por ligeiras opacidades em vidro despolido (C).")
Imagens TAC axiais com algoritmo de alta resolução do tórax (B, D) e imagens TAC axiais de alta resolução (A, C): algumas lucências visíveis (círculos azuis) de pequenas dimensões, semelhantes umas às outras e distribuídas ao longo dos pulmões, bem definidas com uma parede discernível, algumas delas rodeadas por ligeiras opacidades em vidro despolido (C).
 e coronal (B) mostram baixa atenuação de massas císticas retroperitoneal tubulares, que são consistentes com a dilatação dos vasos linfáticos abdominais (seta azul). Linfonodo para-aórtico aumentado (seta vermelha).")
O quadro de linfangioleiomioma ganglionar levanta a possibilidade desses nódulos pulmonares com diferenciação muscular lisa serem uma manifestação pré-clínica de LAM pulmonar ou um envolvimento pulmonar subclínico em pacientes com doença extrapulmonar.
Responsabilidades éticasProteção de pessoas e animaisOs autores declaram que para esta investigação não se realizaram experiências em seres humanos e/ou animais.
Confidencialidade dos dados. Os autores declaram ter seguido os protocolos de seu centro de trabalho acerca da publicação dos dados de pacientes e que todos os pacientes incluídos no estudo receberam informações suficientes e deram o seu consentimento informado por escrito para participar nesse estudo.
Direito à privacidade e consentimento escritoOs autores declaram que não aparecem dados de pacientes neste artigo.
Conflito de interessesOs possíveis conflitos de interesse para cada autor são divulgados.


