

Chronic granulomatous disease associated with common variable immunodeficiency (GD-CVID), although well documented, is rare. Granulomatous lesions can affect several organs and are histologically indistinguishable from sarcoidosis.
Clinical casesCase 1: A 39-year-old male patient with CVID, asymptomatic although with thrombocytopenia and mediastinal-hilar adenopathies. GD-CVID was diagnosed by bone marrow biopsy. Progressive clinical and radiological improvement was obtained with corticotherapy.
Case 2: A 38-year-old male patient with CVID, suffered from asthenia, anorexia, myalgia, lower limbs edemas, and dry cough. He had mediastinal and bilateral hilar adenopathies within which biopsy revealed non-necrotizing granulomatous infiltrate. A spontaneous resolution was detected after 9 months of evolution.
ConclusionGD-CVID is rare and can mimetize other pathologies, namely, sarcoidosis; it should therefore be publicized and discussed so that it becomes a general clinical knowledge.
A doença granulomatosa crónica associada à imunodeficiência comum variável (DG-IDCV), apesar de bem documentada, é rara. As lesões granulomatosas podem afectar vários orgãos e são histologicamente indistinguíveis daquelas que caracterizam a sarcoidose.
Casos clínicosCaso 1: Doente do sexo masculino, 39 anos, com diagnóstico de imunodeficiência comum variável, clinicamente assintomático, com trombocitopenia e adenopatias pré-traqueais e hilares de novo. O diagnóstico de DG-IDCV foi obtido por biópsia de medula óssea. Iniciou tratamento com corticoterapia com melhoria clínica e radiológica progressiva.
Caso 2: Doente do sexo masculino, 38 anos, com diagnóstico de imunodeficiência comum variável, com queixas de astenia, anorexia, mialgias, edemas dos membros inferiores e tosse seca. Adenopatias mediastínicas e hilares bilaterais cuja biopsia revelou infiltrado granulomatoso não necrotizante. Observou-se uma resolução espontânea após nove meses de evolução.
ConclusãoA doença granulomatosa crónica associada a imunodeficiência comum variável é rara. Dado que esta entidade pode mimetizar outras patologias, nomeadamente a sarcoidose, os casos clínicos que se referem à mesma devem ser publicados e discutidos para conhecimento clínico geral.
Common variable immunodeficiency (CVID) is a rare primary immunodeficiency syndrome characterized by impaired B-cell differentiation with inaccurate immunoglobulin production.1 It is defined by markedly reduced serum concentrations of IgG, combined with low levels of IgA and/or IgM, poor or absent response to immunizations in a patient without any other signs of immunodeficiency. The prevalence in Europe is estimated to be around 1:50,000–1:200,000.2 The etiology is still unknown, but it appears to result from a variety of gene defects (e.g., ICOS, TACI, BAFF-R, CD19, CD20, CD21, and CD81), most of which are sporadic rather than familiar.2–4
Although rare, systemic granulomatous disease related to CVID is well documented (>50 case reports). Granulomatous non-caseous lesions can be detected in the lung, liver, spleen, gastrointestinal tract, lymph nodes, skin and eye in almost 10% of the patients with CVID.2,5 Some of them also show clinical manifestations similar to sarcoidosis, with symptoms like dyspnea, persistent cough, asthenia, anorexia and arthralgia.6,7 Granulomatous lesions in CVID may not only affect the same organs as sarcoidosis but also be histologically indistinguishable.8–11 As a consequence, sarcoidosis is often incorrectly diagnosed in GD-CVID patients, with GD-CVID considered as a distinguished entity.12–15
The authors present two clinical cases of CVID with systemic involvement by non- caseous granulomas.
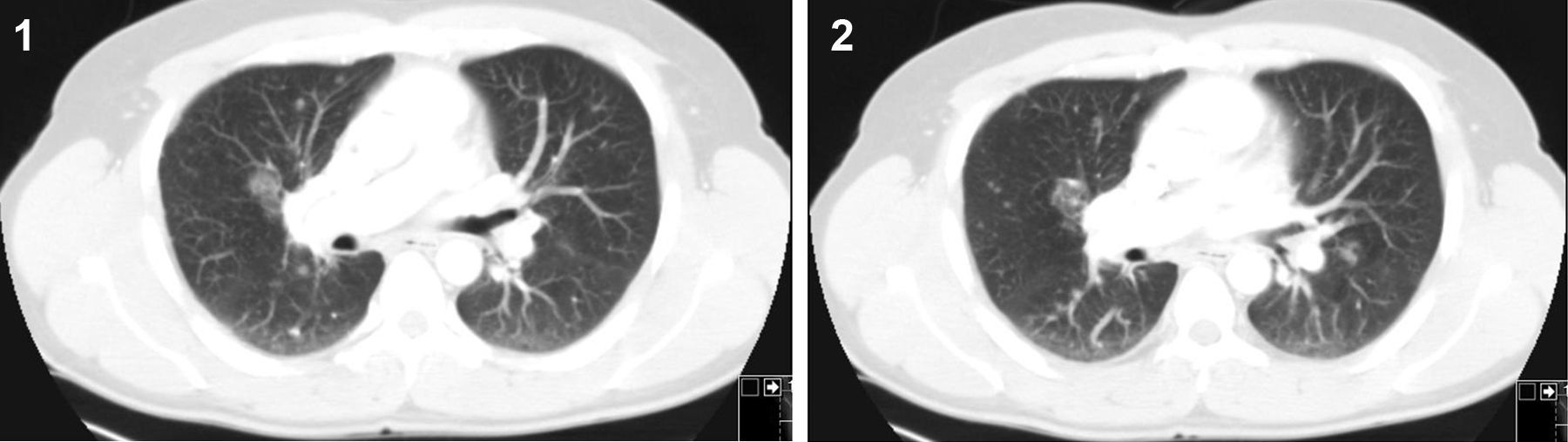
Clinical cases1st caseA 39-year-old male, mechanic engineer, non-smoker, had been diagnosed with CVID 4 years ago, under treatment with intravenous IgG. He had previously had several respiratory infections since childhood. In November 2009, after a routine chest X-ray with mediastinum enlargement, a thoracic CT scan was performed and numerous adenopathies in the prevascular, pretracheal as well as in the right pulmonary hilum were observed. Additionally, diffuse peribronchovascular micronodules were detected in lung parenchyma (Fig. 1 and 2). The patient had no relevant clinical complaints or alterations at physical examination. Besides hypogammaglobulinemia (IgG 287mg/dL; IgM 8mg/dL; IgA 6mg/dL) and slight thrombocytopenia (platelets 127×103μL), no other serum related alterations were noticed. Lung function tests revealed a mild obstructive pattern. A bronchoscopy with bronchoalveolar lavage (BAL) was performed. It showed lymphocytic alveolitis (28%) with a slight increase in CD4/CD8 ratio (2.2), and no microorganism was isolated or malignant cell detected. A bone marrow biopsy was also performed, which showed non-caseous granulomas. There were no clinical or imagiological indications of other organ involvement. At this stage, the patient was started on oral corticotherapy which led to progressive clinical and radiological improvement ending in total disease resolution which continued after the patient come off medication.
2nd case
A 38-year-old male nurse, a non-smoker, had been diagnosed with CVID a year earlier, treated with intravenous IgG. Previously, he had had several respiratory infections since childhood. In February 2010, he began to suffer from asthenia, anorexia, and erythema nodosum. A thoracic CT scan showed mediastinal and bilateral symmetrical hilar adenopathies. Lung function tests were normal. Bronchoscopy with BAL revealed lymphocytosis (35%) with a slight increase in CD4/CD8 ratio (2.6) and no microorganism or malignant cells were detected. Endobronchial ultrasound biopsy showed non-necrotizing granulomatous infiltration. There were no signs of extrathoracic involvement. After diagnosis, he was prescribed symptomatic therapy only (antitussives and anti-inflammatories). After 6 months, there was a spontaneous resolution of the disease, that is to say, the patient became asymptomatic and no imagiologic alterations or signs of recurrence have been detected so far.
DiscussionCVID is a heterogeneous disease, characterized by recurrent infections, particularly bacterial infections often occurring in the respiratory or gastrointestinal tract, autoimmunity phenomena, predisposition to malignant diseases, and granulomatous inflammation (GD-CVID).5,16–18 Although GD-CVID is well documented, the etiology is still unknown and treatment undefined.18
The link between CVID and the granulomatous disease seems logical according to the immunological mechanisms that are implied in the CVID pathophysiology. However, the absence of granulomatous disease in other immunodeficiencies (as hypogammaglobulinemia related to X) suggests that GD-CVID is not caused by recurrent/persistent infections.6 A recent hypothesis is that high levels of tumor necrosis factor (TNF) might be related to the development of granulomas in these patients.6,12
Non-caseous granulomatous lesions that affect CVID patients are histologically indistinguishable from those of sarcoidosis. The patient's age and sex, diminished cellular immunity, the systemic involvement, and high levels of angiotensin-converting enzyme are common features for the two pathologies, so a previous story of recurrent/persistent bacterial infections should raise suspicion of CVID.6,7 However, in a case–control study comparing GD-CVID patients and patients with pulmonary sarcoidosis, certain differences were detected, such as a random versus perilymphatic micronodular distribution on lung CT scan in GD-CVID patients, besides the higher frequency of consolidations with air bronchogram, bronchiectasis, or halo signs. Biological data showed that BAL CD4/CD8 ratio was significantly lower in GD-CVID. There is a general agreement that the presence of lung involvement is associated with a bad prognosis in CVID, including when a GD-CVID occurs. However Bouvry et al. in their case–control study did not find GD to be a critical determinant of prognosis but found a worse outcome to be linked to the classical complications of CVID such as lymphoma, infection or obstructive lung disease secondary to bronchiectasis.1
The clinical presentations of the two reported clinical cases, asymptomatic mediastinal and hilar adenopathies and erythema nodosum, are among the most frequent presentations of sarcoidosis. In addition CVID was already known in both patients; however GD-CVID can, although rarely, be the presentation of the disease. The random distribution of the lung micronodules in the first patient and the slight BAL CD4/CD8 increase are in line with previous reports. However, the therapeutic strategy toward this granulomatous disease has diverged. Although corticotherapy is not universally accepted as a mandatory treatment,12,15 it was prescribed for the first patient because of the systemic involvement, namely, the cytopenia secondary to the bone marrow involvement. The second patient did not receive any therapy directed at the GD. However, both of them improved in clinical, analytical, and radiological terms, which is in line with the favorable evolution reported by Bouvry et al.1 The appropriate treatment for GD-CVID patients has not yet been established.6,7,12,15
There are no protocols regarding diagnosis, treatment and follow-up, nor prognosis for GD-CVID patients. Many of these patients are followed individually and in a non-standardized way. All clinicians should be familiar with this pathology because so many organs and systems can be affected.7,8,17
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Conflicts of interestThe authors have no conflicts of interest to declare.


