






Pulmonary hypertension (PH) is a heterogeneous, debilitating condition with highly relevant impact on functional capacity, quality of life, and life-expectancy.
ObjectivesThis study aims to provide long-term data on the Portuguese PH population, by characterising the clinical presentation, evolution, and outcomes of PH patients in a specialised referral centre.
MethodsRetrospective analysis of a cohort of 101 patients with pre-capillary PH (pcPH) referenced to an expert tertiary care referral centre in northern Portugal from 2002 to 2013. Diagnosis was confirmed by right heart catheterisation (RHC). PH classification followed consensus criteria from the 5th World Symposium in Nice, 2013.
ResultsThe most frequent causes of pcPH were Group 1 PH – pulmonary arterial hypertension (PAH) (54.4%) and Group 4 PH – Chronic thromboembolic pulmonary hypertension (CTEPH) (25.7%); importantly, 17.8% of patients presented PH associated with multiple aetiologies. Targeted therapy was used in 91.1% of patients (48.5% combination therapy). 1-, 3-, and 5-year survival was estimated at 86.6%, 76.7%, and 64.1%, respectively. Survival was significantly better for those ≤40 years old (10.5 vs. 6.4 years; P=0.003) and for women with I/HPAH (9.3 vs. 4.5 years; P=0.039).
ConclusionsThis study provides long-term, real-world data for the management of PAH and CTEPH in Portugal and demonstrates the importance of dedicated electronic medical records and well defined clinical management protocols for better patient outcomes. Patients presented mostly with intermediate or high risk of mortality, which suggests delayed diagnosis and highlights the need to increase awareness among clinicians.
Pulmonary hypertension (PH) is an heterogeneous condition associated with various underlying disorders, which is defined as at rest mean pulmonary arterial pressure (mPAP) ≥25mmHg confirmed by right heart catheterisation (RHC).1 The pathophysiological processes associated with the development of PH are complex and more than likely multifactorial, why explains why several types of classification have been proposed over the years. The most recent international consensus from the 5th World Symposium held in Nice in 2013, classifies PH according to five general groups of aetiologies.2 Group 1 PH refers to pulmonary arterial hypertension (PAH) and includes idiopathic pulmonary arterial hypertension (IPAH), heritable pulmonary arterial hypertension (HPAH), and drugs and toxin induced; PH associated with diseases such as connective tissue disease (CTD), HIV infection, portal hypertension, congenital heart disease (CHD) and schistosomiasis are also included in Group 1. Group 1′ and Group 1″ refer to pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis and persistent pulmonary hypertension of the newborn, respectively. Group 2 PH includes PH due to left heart disease (LHD). Group 3 refers to PH due to lung diseases or hypoxia, such as chronic obstructive pulmonary disease (COPD) or interstitial lung disease. Group 4 PH includes chronic thromboembolic pulmonary hypertension (CTEPH) and other pulmonary artery obstructions. Group 5 refers to PH with unclear and/or multifactorial mechanisms.
Group 1 (PAH) aetiologies, except schistosomiasis associated PAH, are considered rare diseases; IPAH being an exclusion diagnosis, is the most studied form of PAH and the model for clinical management of PAH forms which are indicated for targeted therapy.3,4 Treatment of PH involves both conventional, symptom-based therapy and targeted therapy, which is indicated for specific PH aetiologies. Conventional treatment involves the use of digoxin to improve right ventricular function, diuretics to reduce peripheral oedema, supplemental oxygen, and in specific cases anticoagulants.3 Calcium channel blockers (CCBs) can be used to lower PAP, but their use is restricted to a small percentage of patients (3–5%) showing positive response to acute pulmonary vasodilator (ARVT) challenge.5,6 Targeted therapy includes the use of endothelin-1 receptor antagonists (ERA),7–9 phosphodiesterase-5 inhibitors (PDE-5I),10,11 soluble guanylate cyclase (sGC) stimulators,12,13 and prostacyclin analogues or receptor agonists,14–19 surgical treatment, like lung or heart-lung transplantation is reserved to refractory cases of PAH; pulmonary angioplasty and pulmonary endarterectomy is reserved to CTEPH patients.
The proliferation of studies assessing long-term prognosis of PH has helped identify considerably different patients and disease characteristics both over time and for populations in different geographical regions.20 These findings suggest the need for specific regional data, to fully characterise local disease populations, inform clinical practice, and to help define local/regional political strategies.
In Portugal, a national PH registry has been, but given its recent implementation, only short-term data have been published.21 Recently, another study characterised the survival over a longer follow-up period but the sample size remained relatively small (n=66).22
This study aims to provide long-term data for the Portuguese PH population, by characterising the clinical presentation, evolution, and outcomes of PH patients in a specialised referral centre in Portugal.
Materials and methodsStudy populationWe conducted a retrospective analysis of a cohort of PH patients referenced to an expert tertiary care referral centre in northern Portugal (Pulmonary Vascular Disease Unit, Centro Hospitalar do Porto – Hospital de Santo António, Porto, Portugal) from 2002 to 2013. At this centre, patients followed a defined protocol for the clinical management of PH, which was adjusted to the applicable national23 and international guidelines during the period of the study. The protocol specified mandatory clinical assessments, which were prospectively collected in dedicated PH software developed by the centre (PAHTool®, Inovultus Lda, Santa Maria da Feira, Portugal).
PH was confirmed by right heart catheterisation (RHC), with a mean pulmonary artery pressure (mPAP) ≥25mmHg; pulmonary arterial wedge pressure (PAWP) ≤15mmHg was used to define pre-capillary PH. For the purposes of this study, clinical classification of PH followed standard criteria according to the consensus from the 5th World Symposium in Nice, 2013. Patients with left heart disease (LHD) (Group 2 PH) were not included in this study, due to the significantly different pathophysiology, treatment approaches, and prognosis.
The study was approved by the ethics committee of Hospital de Santo António, Porto, Portugal. All patients provided their written informed consent prior to enrolment in the study.
Medical carePatients received standard medical care throughout the period of the study. Treatment was prescribed by the accompanying physician based on national and international guidelines applicable at time of the study (as defined in the protocol implemented at the centre) and local treatment availability. Overall, all patients received standard conventional treatment when clinically indicated, including anticoagulants, diuretics, digoxin, oxygen supplementation, and high dose calcium channel blockers (CCBs) (if they were AVRT responders). Selected patients received molecular targeted therapy in addition to conventional therapy, including endothelin-1 receptor antagonists (ERA), phosphodiesterase-5 inhibitors (PDE-5I), and prostacyclin analogues.
AssessmentsDemographic characteristics (gender and age) and clinical characteristics (PH aetiology, symptoms, WHO functional class, 6-min walking test (6MWT), N-terminal pro brain natriuretic peptide (NT-proBNP), and haemodynamic parameters) were collected at baseline. According to the protocol implemented at the centre, patients attended, routinely, 3–4 visits per year or if they had any sign of deterioration. During follow-up the following assessments were considered mandatory: type of treatment administered, clinical evaluation focused on signs of deterioration (like heart failure or syncope), WHO functional class, 6MWT, and NT-proBNP; and yearly haemodynamic re-evaluation. Survival was established based on the electronic medical records (EMR).
Statistical methodsDemographic and clinical variables were summarised with descriptive statistics. Categorical variables were summarised as absolute frequency and percentage, whereas continuous variables were summarised as mean and standard deviation (SD). Student's t, Wilcoxon's, Fisher's exact, or chi-square tests were used to conduct paired/independent univariate/bivariate analysis as appropriate. Cumulative survival was estimated using the Kaplan–Meier method. Patients were censored at the end of the study, except for those who underwent lung transplantation, censored at the time of transplantation. Differences between the survival curves (according to baseline characteristics and disease aetiology) were analysed using the log-rank test. A 5% significance level was employed for all analyses. For the purpose of subgroup analysis patients with multiple aetiology were excluded from any subgroup analysis, except those for whom CTEPH was considered their main diagnosis.
Data was retrieved from PAHTool®. Statistical analysis was conducted using IBM SPSS Statistics for Windows, Version 21.0 (IBM Corp., Armonk, NY, USA).
ResultsStudy population and baseline characteristicsDuring the enrolment period of the study, a total of 211 patients with suspected PH were referenced to the Pulmonary Vascular Disease Unit (PDVU). RHC confirmed PH in 120 patients, of which, 19 were excluded from the analysis (10 patients lost to follow-up and 9 patients with exclusive left heart disease), leading to a final study cohort of 101 patients with pcPH. The majority of patients represented incident cases (n=81, 80.2%), however, 20 (19.8%) patients had a prior diagnosis of congenital heart diseases before referral, having started specific therapy at admission to the centre. Fig. 1 presents the patient disposition in the study cohort.

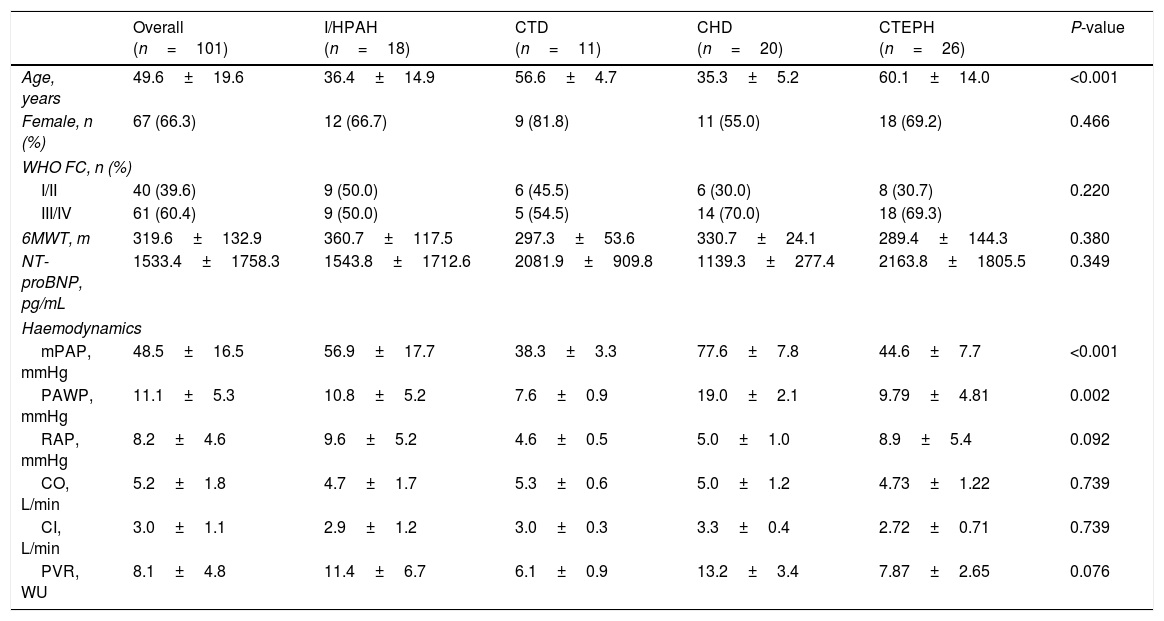
The mean±SD follow-up time in the overall study population was 3.8±2.7 years. Table 1 presents the baseline demographic and clinical characteristics of the study population. Approximately 66.3% of patients were female (3:1 ratio) and the mean±SD age at baseline was 49.6±19.6 years. Most participants showed moderate to severe disease manifestations at baseline, with 60.4% of patients presenting in WHO functional classes (FC) III or IV, with a mean NT-proBNP level of 1533.4±1758.3pg/mL, and walking a mean distance of 319.6±132.9m on 6MWT. Haemodynamically, patients showed increased mPAP of 48.5±16.5mmHg, increased PVR of 8.1±4.8 wood units, increased RAP 11.1±5.3mmHg, and normal CI of 3.0±1.1L/min.
Baseline demographic and clinical characteristics.
| Overall (n=101) | I/HPAH (n=18) | CTD (n=11) | CHD (n=20) | CTEPH (n=26) | P-value | |
|---|---|---|---|---|---|---|
| Age, years | 49.6±19.6 | 36.4±14.9 | 56.6±4.7 | 35.3±5.2 | 60.1±14.0 | <0.001 |
| Female, n (%) | 67 (66.3) | 12 (66.7) | 9 (81.8) | 11 (55.0) | 18 (69.2) | 0.466 |
| WHO FC, n (%) | ||||||
| I/II | 40 (39.6) | 9 (50.0) | 6 (45.5) | 6 (30.0) | 8 (30.7) | 0.220 |
| III/IV | 61 (60.4) | 9 (50.0) | 5 (54.5) | 14 (70.0) | 18 (69.3) | |
| 6MWT, m | 319.6±132.9 | 360.7±117.5 | 297.3±53.6 | 330.7±24.1 | 289.4±144.3 | 0.380 |
| NT-proBNP, pg/mL | 1533.4±1758.3 | 1543.8±1712.6 | 2081.9±909.8 | 1139.3±277.4 | 2163.8±1805.5 | 0.349 |
| Haemodynamics | ||||||
| mPAP, mmHg | 48.5±16.5 | 56.9±17.7 | 38.3±3.3 | 77.6±7.8 | 44.6±7.7 | <0.001 |
| PAWP, mmHg | 11.1±5.3 | 10.8±5.2 | 7.6±0.9 | 19.0±2.1 | 9.79±4.81 | 0.002 |
| RAP, mmHg | 8.2±4.6 | 9.6±5.2 | 4.6±0.5 | 5.0±1.0 | 8.9±5.4 | 0.092 |
| CO, L/min | 5.2±1.8 | 4.7±1.7 | 5.3±0.6 | 5.0±1.2 | 4.73±1.22 | 0.739 |
| CI, L/min | 3.0±1.1 | 2.9±1.2 | 3.0±0.3 | 3.3±0.4 | 2.72±0.71 | 0.739 |
| PVR, WU | 8.1±4.8 | 11.4±6.7 | 6.1±0.9 | 13.2±3.4 | 7.87±2.65 | 0.076 |
Results are presented as mean±SD, except when otherwise indicated.
IPAH: idiopathic pulmonary arterial hypertension; HPAH: heritable pulmonary arterial hypertension; CTD: connective tissue disease; CHD: congenital heart disease; CTEPH: chronic thromboembolic pulmonary hypertension; WHO FC: World Health Organization functional class; 6MWD: 6-min walk distance; NT-proBNP: N-terminal pro brain natriuretic peptide; mPAP: mean pulmonary artery pressure; PCWP: pulmonary capillary wedge pressure; RAP: right atrial pressure; CO: cardiac output; CI: cardiac index; PVR: pulmonary vascular resistance.
The most frequent causes of PH were Group 1 PH – PAH (54.4%) and Group 4 PH – CTEPH (25.7%); from the PAH subgroup, CHD (36.3%) was the most frequent, followed by I/HPAH (32.7%), and CTD (20.0%); importantly, 17.8% of patients presented PH of multiple aetiology.
Concerning the most frequent PH subgroups, I/HPAH and CHD patients were younger (P<0.001); for all subgroups of aetiologies most patients presented with WHO FC III/IV, but patients with CHD and CTEPH showed significantly (P<0.001) worst functional capacity at baseline.
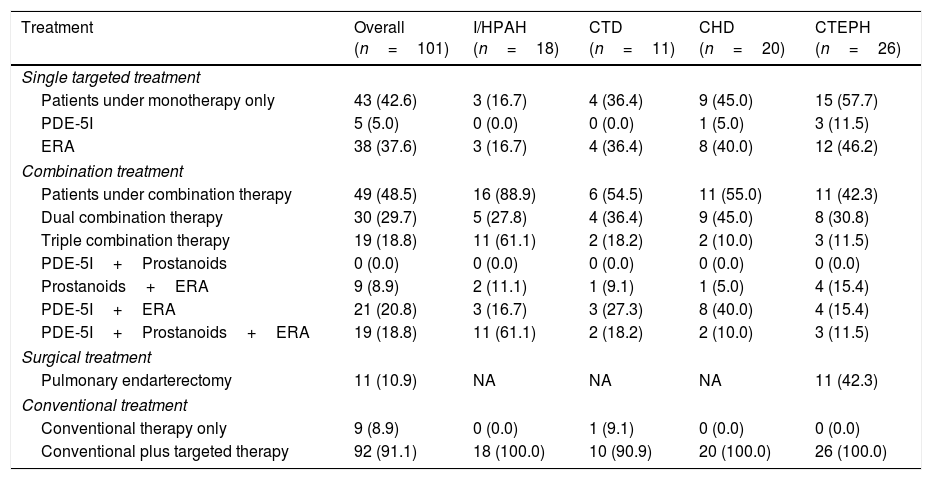
Treatment and clinical evolutionTable 2 shows PH treatment used at the last follow-up visit. All patients received conventional treatment during the period of the study, with 8.9% of those receiving conventional therapy only. Targeted therapy was used with 91.1% of participants in addition to conventional therapy. 4 (6.8%) I/HPAH patients were AVRT responders, but only 2 were long-term responders, being treated with high doses of CCBs only.
Medical and surgical treatment at the last follow-up visit.
| Treatment | Overall (n=101) | I/HPAH (n=18) | CTD (n=11) | CHD (n=20) | CTEPH (n=26) |
|---|---|---|---|---|---|
| Single targeted treatment | |||||
| Patients under monotherapy only | 43 (42.6) | 3 (16.7) | 4 (36.4) | 9 (45.0) | 15 (57.7) |
| PDE-5I | 5 (5.0) | 0 (0.0) | 0 (0.0) | 1 (5.0) | 3 (11.5) |
| ERA | 38 (37.6) | 3 (16.7) | 4 (36.4) | 8 (40.0) | 12 (46.2) |
| Combination treatment | |||||
| Patients under combination therapy | 49 (48.5) | 16 (88.9) | 6 (54.5) | 11 (55.0) | 11 (42.3) |
| Dual combination therapy | 30 (29.7) | 5 (27.8) | 4 (36.4) | 9 (45.0) | 8 (30.8) |
| Triple combination therapy | 19 (18.8) | 11 (61.1) | 2 (18.2) | 2 (10.0) | 3 (11.5) |
| PDE-5I+Prostanoids | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Prostanoids+ERA | 9 (8.9) | 2 (11.1) | 1 (9.1) | 1 (5.0) | 4 (15.4) |
| PDE-5I+ERA | 21 (20.8) | 3 (16.7) | 3 (27.3) | 8 (40.0) | 4 (15.4) |
| PDE-5I+Prostanoids+ERA | 19 (18.8) | 11 (61.1) | 2 (18.2) | 2 (10.0) | 3 (11.5) |
| Surgical treatment | |||||
| Pulmonary endarterectomy | 11 (10.9) | NA | NA | NA | 11 (42.3) |
| Conventional treatment | |||||
| Conventional therapy only | 9 (8.9) | 0 (0.0) | 1 (9.1) | 0 (0.0) | 0 (0.0) |
| Conventional plus targeted therapy | 92 (91.1) | 18 (100.0) | 10 (90.9) | 20 (100.0) | 26 (100.0) |
Results are presented as absolute frequency (percentage).
IPAH: idiopathic pulmonary arterial hypertension; HPAH: heritable pulmonary arterial hypertension; CTD: connective tissue disease; CHD: congenital heart disease; CTEPH: chronic thromboembolic pulmonary hypertension; PDE-5I: phosphodiesterase-5 inhibitors; ERA: endothelin-1 receptor antagonists; NA: not applicable.
Single targeted therapy was used in 42.6% of patients, dual combination therapy in 29.7%, and triple combination therapy in 18.8%. 42.3% of CTEPH patients underwent pulmonary endarterectomy during the course of the study. The majority of patients with I/HPAH were under combination therapy (88.9%), with 61.1% under triple therapy. Most patients with CTD (54.4%) and CHD (55.0%) were under combination therapy, and dual therapy was the most frequent type of treatment (36.4% and 45.0%, respectively). Patients with non-operable CTEPH or with residual persistent PH were mostly under monotherapy (57.7%), particularly with ERAs (46.2%).
During patient follow-up, functional capacity improved significantly (P<0.003) from first to last visit for the overall study population, as illustrated in Fig. 2. Mean 6MWD significantly improved in the overall population (P=0.003) and in the subgroup of I/HPAH patients (P=0.011). CHD subgroup was the only one to show a significant (P=0.040) improvement in NT-proBNP levels (Table 3).

6MWD and NT-proBNP evolution from first to last visit.
| Overall (n=101) | I/HPAH (n=18) | CTD (n=11) | CHD (n=20) | CTEPH (n=26) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| First visit | Last visit | First visit | Last visit | First visit | Last visit | First visit | Last visit | First visit | Last visit | |
| 6MWD, m | 319.6±132.9 | 357.7±162.1** | 360.7±117.5 | 463.3±163.1 | 297.3±53.6 | 282.5±163.8 | 330.7±24.1 | 319.8±164.8 | 289.4±144 | 337.2±156.2 |
| NT-proBNP, pg/mL | 1533.4±1758.3 | 1963.9±3627.2 | 1543.8±1712.5 | 1583.2±3761.9 | 2081.9±909.8 | 3392.8±3732.1 | 1139.3±277.4 | 1815.9±3671.7* | 2163.8±1805.5 | 2960.1±5098.3 |
Results are presented as mean±SD.
6MWD: 6-min walk distance; NT-proBNP: N-terminal pro brain natriuretic peptide.
There was a significant reduction in mPAP in the overall population (P=0.002) and in the subgroup of I/HPAH patients (P=0.008). PVR significantly improved in the overall population (P=0.008) and in the subgroup of I/HPAH patients (P=0.008). No significant changes were observed in RAP and CI for either the overall population or subgroup analyses.
SurvivalDuring the follow-up period, a total of 28 (27.7%) patients died of PH-related causes; 10.7% of deaths occurred in patients with I/HPAH, 25.0% with CTEPH, 10.7% with CTD, and 25.0% with CHD. Patients with WHO FC III or IV at baseline represented 67.9% of deaths. Median survival time from diagnosis by RHC for the 28 deaths was 3.1 years. At the time of death 35.7% of patients were under monotherapy, 39.3% under dual therapy, 17.9% under triple therapy, and only 2 patients (7.1%) were under exclusive conventional therapy.
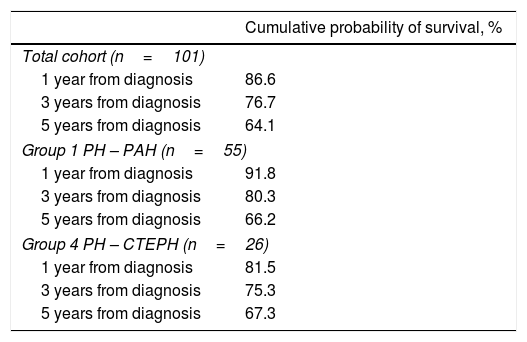
Table 4 presents survival estimates for the overall study cohort and specific PH aetiologies. For the overall study cohort, 1-, 3-, and 5-year survival was estimated at 86.6%, 76.7%, and 64.1%, respectively. Fig. 3 shows the Kaplan–Meier survival curve for the overall cohort and for patients with PAH vs. CTEPH. Fig. 4 shows specific Kaplan–Meier survival curves for subgroups of PAH (A) and according to CTEPH treatment (B).
Kaplan–Meier survival estimates.
| Cumulative probability of survival, % | |
|---|---|
| Total cohort (n=101) | |
| 1 year from diagnosis | 86.6 |
| 3 years from diagnosis | 76.7 |
| 5 years from diagnosis | 64.1 |
| Group 1 PH – PAH (n=55) | |
| 1 year from diagnosis | 91.8 |
| 3 years from diagnosis | 80.3 |
| 5 years from diagnosis | 66.2 |
| Group 4 PH – CTEPH (n=26) | |
| 1 year from diagnosis | 81.5 |
| 3 years from diagnosis | 75.3 |
| 5 years from diagnosis | 67.3 |
PH: pulmonary hypertension; PAH: pulmonary arterial hypertension; CTEPH: chronic thromboembolic pulmonary hypertension. Survival estimates for subgroups of PH were not calculated due to limitations introduced by reduced sample size and variable follow-up times in the subgroups.
 and patients with CTEPH (Group 4 PH).")
 subgroup of PAH and (B) CTEPH treatment.")
Survival was significantly better for those ≤40 years old (10.5 vs. 6.4 years; P=0.003) and in women with I/HPAH compared with men (9.3 vs. 4.5 years; P=0.039); no other significant differences in survival were observed for gender, age, PH aetiology, and functional capacity analyses for the overall population and subgroups of aetiologies.
DiscussionThis study provides long-term data for patient phenotypes, clinical evolution, and survival in PAH and CTEPH of a Portuguese PH population. These results build on previous findings21,24 and together they characterise the impact of this life-threatening disease in Portugal. The present study enrolled only patients with pcPH due to the substantially different disease characteristics and treatment approaches that could bias the results and ultimately hinder comparisons with other cohorts published over the years. Unlike previous studies, here we included patients with all types of PH, except for left heart disease, to provide a more accurate picture of pcPH as a whole. Left heart disease was the only PH group excluded from the analysis.
The PH dedicated software developed by the centre allowed the implementation of a mandatory case report form (CRF) with automatic alerts to avoid missing data in our clinical records. This methodology allowed us to build a true real-world cohort of PH patients from our region and following the most recent Nice recommendations.25 This is particularly relevant because PAHTool® is currently licenced for implementation in several PH centres worldwide, and its widespread use is envisioned to allow the generation of relevant real-world data, which is badly needed in the context of this non-frequent condition but rapidly evolving field.
According to existing registries, some aetiologies are underrepresented in our cohort (particularly drugs and toxins induced PH, HIV and portal hypertension associated PH), which is most likely due to low physician awareness about PH.21,26–36 CHD (36.3% of PAH group) contributed with a comparatively higher proportion of cases, which can be explained by the past low levels of detection and correction of heart defects in infancy in our country and by the fact that our centre was for a long time the only one to provide targeted therapy for PAH in the region. CTEPH was the most frequent aetiology after PAH, confirming the high prevalence of this frequently forgotten condition and in line with studies that included this subgroup of patients.21,34,37–39
Comparing our data with the most important registries in the field, we found that CTEPH age at diagnosis in our population is consistent with these registries38–40; although the mean age at diagnosis for our overall population is similar to the majority of PAH registries26,29,32 the mean age for I/HPAH is clearly lower and near the pioneer registries35,41 and those coming from the developing world,42,43 but still in line with national data.21 Female gender (66%) predominance is also in line with the majority of PH registries and cohorts.21,26–35
Baseline clinical characteristics at presentation indicate some delay in diagnosis, with most patients presenting with intermediate or high risk of mortality indicated by high NYHA FC, low 6MWD and high NT-proBNP according to risk assessment guidelines3; increased RAP (11.1±5.3mmHg) and PVR (8.1±4.8WU) are also consistent with these findings. The proportion of patients presenting with intermediate or high risk explains why the great majority of patients were treated with combination therapy: I/HPAH (88.9%), CTD (54.5%) and CHD (55.0%). These findings are, however, above what has been reported in recent European studies, as well as national data.21,30,32,34,44 There is a particularly high proportion of I/HPAH (61.1%) patients under triple therapy, which is probably the result of the close follow-up adopted and continuous risk evaluation with early step up of the therapy. ERAs and PDE-5I were the most widely used drugs, which is in accordance with previous reports.21,30,32,34,44 Still, they were used far more frequently in combination than what has been reported, which is in line with the most recent recommendations for the early use of sequential or upfront double combination therapy.3,45
Although pulmonary endarterectomy surgery is not routinely available in our country, patients are fully reimbursed by the Portuguese Public National Health Service if surgery is performed abroad. Despite limitations associated with the need for a cross-border, high risk procedure, 42% of CTEPH patients had the operation in a foreign centre, thanks to a protocol for surgical treatment of PH established in 2000. Non-operable patients, patients refusing pulmonary endarterectomy, or patients with residual persistent PH after pulmonary endarterectomy were predominantly treated with monotherapy (57.7%) and specially with ERAs (46.2%), as per previous studies.39,40
In terms of clinical outcomes during the period of the study, WHO FC, 6MWD, mPAP and PVR significantly improved for the overall population, which is in line with the accumulating evidence of substantial gains in long-term prognosis obtained over recent decades, following the introduction of several therapeutic alternatives.3,30,32,34
Although usual bias for survival estimates were eliminated in this study (including mixed population of incident/prevalent cases and “immortal” bias), survival estimates should still be considered with caution since there were substantial changes in treatment strategies during the course of the study. Nonetheless, survival estimates found for the total cohort and PAH follow the trends of the most relevant international registries26–28,30,32,35; the 1-year survival among CTEPH patients (81.5%) was considerably below estimates from other studies (88–97%),37–40 which could be connected to difficulties of access to PEA surgery.
This study had several limitations. First, its single-centre nature might affect the representativeness of the findings for the overall Portuguese population, however, the study was conducted at one of the largest PH centres in the country, serving the northern region, with an ample and diverse group of patients currently being followed. Second, the relatively small sample size impaired the ability to perform more in-depth statistical analysis such as predictors of survival, however, in the context of a non-frequent disease the data provided by this study gives highly relevant insight to inform clinical practice. Third, the enrolment period for this study was long, which is associated with highly variable follow-up times and variable treatment approaches over time. Still, the low incidence of PH and the country/region dimension make enrolment over reduced periods difficult.
ConclusionsThis study provides long-term, real-world data for the management of PH in Portugal. It also demonstrates the potential of a dedicated information system for PAH in generating high-quality real-world data aimed at characterising PH in present clinical practice conditions. Patients presented mostly with intermediate or high risk of mortality which might be indicative of delay in diagnosis and highlights the need to increase awareness and early referral to expert centres for this condition among clinicians.
Conflicts of interestAbílio Reis is co-proprietary of the dedicated pulmonary hypertension software PAHTool®. The remaining authors have no conflicts of interests to disclose.
The authors would like to Tiago Campos (ARC Publishing) for providing medical writing assistance; these services were supported by an unrestricted grant from Actelion Pharmaceuticals.


