

A Fibrose Pulmonar Idiopática (FPI) é a patologia mais comum no subgrupo das pneumonias intersticiais idiopáticas. Apesar de uma grande variabilidade no tipo de evolução clínica, está inexoravelmente associada a um mau prognóstico.
Material e métodosForam identificados doentes com FPI, observados na consulta de doenças pulmonares difusas do Centro Hospitalar de São João, Porto, e revistos os seus parâmetros clínicos, funcionais, radiológicos e do lavado broncoalveolar (LBA). A evolução clínica e sobrevivência foram avaliadas, tendo sido igualmente identificados fatores prognósticos.
ResultadosForam incluídos 81 doentes com uma média de idade de 63,8 anos. Na altura do diagnóstico, as principais alterações funcionais identificadas foram a restrição, redução da difusão pulmonar e da capacidade de exercício. A maioria dos doentes (72.3%) apresentou uma evolução clínica lentamente progressiva. Em 10 doentes (13.2%), foi observada uma evolução rapidamente progressiva e 11 (14.,5%) apresentaram exacerbação aguda. Verificou-se uma associação entre a evolução rapidamente progressiva e uma maior gravidade funcional ao diagnóstico, nomeadamente na Capacidade Vital Forçada (CVF) e Capacidade Pulmonar Total (CPT). A sobrevida mediana foi de 36 meses. Verificou-se uma diferença estatisticamente significativa na sobrevida entre os diferentes grupos de evolução clínica: 41 meses para os doentes com evolução lentamente progressiva e 9 meses para os doentes com evolução rapidamente progressiva. Valores inferiores de CVF, CPT, distância na Prova da Marcha de 6 Minutos (PM6 M) e PaO2 em repouso, bem como o maior grau de neutrofilia no LBA estiveram associados a uma sobrevivência inferior em análise univariada.
ConclusãoA análise deste grupo de doentes com FPI confirma a existência de 2 fenótipos claramente distintos, o de evolução lenta e o de evolução rapidamente progressiva. Estes fenótipos têm uma diferente apresentação clínica e uma história natural da doença claramente distinta, sugerindo a presença de diferentes mecanismos fisiopatológicos, os quais poderão implicar diferentes abordagens terapêuticas.
Idiopathic pulmonary fibrosis (IPF) is the most common disease in the subgroup of idiopathic interstitial pneumonias. It is inevitably associated to a bad prognosis, although assuming a highly variable clinical course.
MethodsPatients with IPF, observed at Interstitial Lung Diseases outpatient clinic of Centro Hospitalar de São João – Porto, Portugal, were identified and clinical, functional, radiological and bronchoalveolar lavage (BAL) parameters were reviewed. Their clinical course and survival were analyzed in order to identify prognostic factors.
ResultsEighty-one patients were included, with a mean age at diagnosis of 63.8 years old. At diagnosis, the main functional abnormalities were restrictive physiology, reduced lung diffusion and exercise capacity impairment. Clinical course was mainly slowly progressive (72.3%). Ten patients (13.2%) had a rapid progression and 11 (14.5%) patients had an acute exacerbation during the course of the disease. IPF's rapid progression was associated to a higher functional impairment at diagnosis, namely in what is related with Forced Vital Capacity (FVC) and Total Lung Capacity (TLC). Median survival was 36 months. A significant difference in survival was observed among different types of clinical course – 41 months for slow progressors and 9 months for rapid progressors. Lower levels of FVC, TLC, 6th minute walk test distance and rest PaO2, and higher BAL neutrophil count were associated with poorer survival in univariate analysis.
ConclusionThe analysis of this group of IPF patients confirms two clearly different phenotypes, slow and rapid progressors. Those phenotypes seem to have different presentations and a remarkably different natural history. These results could mean different physiopathologic pathways, which could implicate different therapeutic approaches.
A Fibrose Pulmonar Idiopática (FPI) é a patologia mais comum no subgrupo das pneumonias intersticiais idiopáticas (PII)1,2. É uma doença associada ao envelhecimento, com apresentação geralmente nas sexta e sétima décadas de vida, e é mais frequente no sexo masculino3–5. Quer a sua etiologia, quer os mecanismos da doença, são ainda largamente desconhecidos3.
O padrão histológico típico da FPI é a pneumonia intersticial usual (UIP)6. Na ausência de biopsia, a FPI pode ser diagnosticada com base nos critérios ATS/ERS – grupo de critérios clínicos, radiológicos e fisiológicos, internacionalmente aceites e validados, nos quais os aspetos típicos na tomografia computorizada torácica de alta resolução (TCAR) têm um papel proeminente3,7.
A história natural da doença é altamente variável, ocorrendo geralmente uma deterioração fisiológica lenta e progressiva. Contudo, nalguns doentes observa-se um declínio rápido da função pulmonar e o óbito ocorre 6 a 12 meses após o diagnóstico8–10. Noutros doentes ainda, observa-se uma exacerbação aguda durante a evolução da doença – agravamento respiratório súbito, com hipoxemia e aparecimento de novos infiltrados radiológicos, sem causa identificável11,12.
Apesar dos diferentes tipos de evolução clínica, a FPI está inevitavelmente associada a um mau prognóstico, com uma sobrevivência mediana de 2-3 anos8,13. Não existe ainda uma terapêutica farmacológica de eficácia comprovada3. Estão já descritos alguns fatores prognósticos, o que tem implicações terapêuticas, nomeadamente no que respeita ao tempo de referenciação para transplante pulmonar, a única terapêutica até ao presente associada a um benefício na sobrevivência14,15. Recentemente, a pirfenidona, um fármaco com propriedades antifibróticas e anti-inflamatórias, foi aprovado pela EMEA (European Medicines Agency) no tratamento da FPI.
O objetivo deste estudo foi a descrição da apresentação clínica e da evolução dos doentes com FPI observados na consulta de doenças pulmonares difusas (DPD), e a análise de fatores relacionados com a sobrevivência e o tipo de evolução.
MétodosForam avaliados os doentes com FPI, observados na consulta de DPD do Hospital de São João, um centro de referência terciário no Porto, Portugal. Em 56 doentes (69,1%), o diagnóstico foi baseado nos critérios ATS/ERS (de acordo com o consenso de 2002)2. Sempre que existiam dúvidas no diagnóstico, ou por alterações atípicos na TCAR, ou por outras características atípicas, os doentes eram referenciados para realização de biópsia pulmonar cirúrgica, que foi efetuada em 25 doentes (30,9%). Aqueles cujo padrão radiológico ou histológico de UIP pudesse ser explicado por outras patologias (nomeadamente doenças do tecido conjuntivo, pneumonite de hipersensibilidade, toxicidade pulmonar a fármacos ou outras DPD cujo estadio final é fibrose pulmonar) foram excluídos.
Os registos clínicos foram analisados retrospetivamente. Os doentes foram caracterizados à data do diagnóstico, do ponto de vista clínico, fisiológico e imagiológico.
Os parâmetros fisiológicos em repouso e no exercício (espirometria, capacidade pulmonar total – CPT, difusão pulmonar do monóxido de carbono – DLCO, prova da marcha dos 6 minutos – PM6M e prova de exercício cardiopulmonar) foram avaliados de acordo com as recomendações da European Respiratory Society (ERS) e American Thoracic Society (ATS)16–20, sendo os resultados expressos como percentagens dos valores normais previstos. Foram excluídos os parâmetros fisiológicos obtidos após qualquer medicação que pudesse eventualmente modificar o curso natural da doença.
Em alguns doentes não foi possível a avaliação da capacidade de exercício devido a um elevado grau de incapacidade ao esforço.
Todos os doentes realizaram TCAR na altura do diagnóstico. Contudo, tratando-se de um estudo retrospetivo, alguns doentes não tinham imagens de TCAR disponíveis para análise. Para a caracterização imagiológica, as imagens da TCAR foram avaliadas por 2 radiologistas, que classificaram a extensão da doença através de um score fibrótico21, o mesmo utilizado na validação do Composite Physiologic Index.
O lavado broncoalveolar (LBA) foi efetuado em regime de ambulatório, sendo de referir que, na altura da realização deste, não havia sinais ou sintomas de infeção respiratória ou de exacerbação da doença. Quer a realização do LBA, quer o seu processamento, obedeceram às recomendações do grupo de trabalho de LBA da ERS22. A contagem celular total e diferencial foi classificada de acordo com os valores propostos por este grupo de trabalho (linfocitose > 15%, neutrofilia > 3%, eosinofilia > 0,5%)22.
A biópsia pulmonar aberta foi realizada sob anestesia geral, sendo a abordagem mais comummente utilizada a toracotomia ântero-lateral através de uma incisão submamária de 4-10cm, incisão esta que permite o acesso a diferentes segmentos e lobos pulmonares, de forma a obter múltiplas amostras de biopsia. O local de realização da biópsia foi escolhido com base na TCAR. As amostras de biopsia foram posteriormente observadas e avaliadas de forma independente por 2 anátomo-patologistas.
Para averiguar a existência de hipertensão pulmonar, todos os doentes realizaram ecocardiograma. Apenas os doentes referenciados para transplante pulmonar foram submetidos a cateterização cardíaca direita.
A evolução clínica foi classificada em evolução lenta, evolução rápida e exacerbação aguda (EA-FPI). A evolução rápida foi diferenciada da evolução lenta por uma menor duração de sintomas prévios ao diagnóstico (<6 meses) e uma deterioração clínica rapidamente progressiva10. A EA-FPI foi definida como um agravamento súbito dos sintomas respiratórios com hipoxemia e aparecimento de infiltrados pulmonares de novo, sem etiologia identificável11.
Nos doentes incluídos neste estudo, a FPI foi diagnosticada entre 2000 e 2010. Foi feito o seguimento da evolução da doença até à data de óbito ou de transplante pulmonar.
Análise estatísticaA normalidade da distribuição de todas as variáveis contínuas foi testada através do teste de Kolmogorov-Sminorv. Quanto aos fatores estudados, foram analisadas as suas percentagens e medianas das covariadas e as diferenças entre os doentes com evoluções lenta e rápida foram averiguadas através dos testes U Mann-Whitney, Qui Quadrado ou Teste Exato de Fisher, conforme apropriado.
A sobrevivência mediana dos doentes foi estimada através das curvas de sobrevivência de Kaplan-Meier, sendo estes censurados à data de óbito ou de transplante pulmonar. Para aumento do poder estatístico do estudo, o seguimento dos doentes foi suspenso aos 48 meses. Para avaliar os fatores prognósticos, os Hazard Ratios (e seus respetivos intervalos de confiança a 95%) foram analisados usando os modelos de regressão univariados de Cox.
Todas as análises estatísticas foram efetuadas com recurso ao SPSS® software v. 18, para um nível de significância estatística de 0,05.
ResultadosCaracterização dos doentes à data do diagnósticoForam incluídos 81 doentes, 56 (69,1%) do sexo masculino e 25 (30,9%) do sexo feminino. A média de idade ao diagnóstico foi de 63,8±10,2 anos. Dois doentes (2,5%) apresentaram FPI familiar. Foi identificada história tabágica em 51,9% (27,5% fumadores e 72,5% ex-fumadores), com uma carga tabágica média de 37,5 unidades maço/ano (UMA). O principal sintoma à apresentação foi a dispneia (96,3%), seguido de tosse (7,9%). O tempo mediano entre o início dos sintomas e a data do diagnóstico foi de 12 meses.
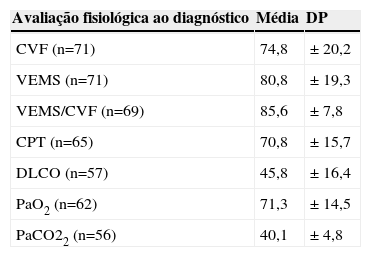
As principais alterações funcionais encontradas foram uma alteração ventilatória restritiva e a redução da DLCO, bem como a redução da capacidade de exercício (tabela 1). A PM6M foi considerada para análise em 40 doentes, com uma média (DP – desvio-padrão) de distância percorrida de 369,6 (149,4) metros e uma média (DP) de saturação mínima de oxigénio de 81,5 (8,1) %. A prova de exercício cardiopulmonar foi considerada para análise em apenas 15 doentes, com uma média (DP) de pico de consumo de oxigénio de 66,6 (18,4) %.
Avaliação fisiológica dos doentes com FPI no momento do diagnóstico
| Avaliação fisiológica ao diagnóstico | Média | DP |
|---|---|---|
| CVF (n=71) | 74,8 | ± 20,2 |
| VEMS (n=71) | 80,8 | ± 19,3 |
| VEMS/CVF (n=69) | 85,6 | ± 7,8 |
| CPT (n=65) | 70,8 | ± 15,7 |
| DLCO (n=57) | 45,8 | ± 16,4 |
| PaO2 (n=62) | 71,3 | ± 14,5 |
| PaCO22 (n=56) | 40,1 | ± 4,8 |
DP: desvio-padrão; PaCO2: pressão arterial de dióxido de carbono; PaO2: pressão arterial de oxigénio; VEMS: volume expirado máximo no 1.° segundo.
O score fibrótico na TCAR foi medido em 42 doentes, com uma média (DP) de 10,8 (2,3).
A prevalência de enfisema foi de 26,4% (14/53 TCAR disponíveis para avaliação). Nos doentes com enfisema, verificou-se uma idade inferior – 61,8 vs. 69,9 anos (p=0,042), uma maior carga tabágica – 28,3 vs. 10,7 UMA (p=0,035), uma menor relação VEMS/CVF – 79 vs. 86,9% (p=0,001) e um maior compromisso na capacidade de difusão – 36,8 vs. 49,9% (p=0,016).
Na abordagem diagnóstica, foi realizado LBA em 56 doentes, identificando-se neutrofilia (média=10,8, DP=2.3) em 52 (92,9%) doentes, eosinofilia (média=5,1, DP=5,0) em 48 (85,7%) doentes e uma linfocitose ligeira em 14 (25%) doentes (média=12, DP=7,8).
Evolução clínicaNo que respeita à abordagem terapêutica, foram seguidas as normas ATS/ERS à data – na ausência de contraindicações, tendo esta consistido em doses baixas de corticosteroides (86,4%) e fármacos imunossupressores (65,4%), preferencialmente azatioprina ou ciclofosfamida. Após a publicação do ensaio clínico IFIGENIA23 em 2005, foi adicionado a este regime terapêutico a N-Acetilcisteína (1800mg/dia) – 51,9%. Até à data de realização deste estudo, a Pirfenidona não estava ainda disponível em Portugal. À data de diagnóstico dos doentes, este fármaco não estava ainda aprovado pela EMEA nem havia centros portugueses a participar nos seus ensaios clínicos.
Após o estabelecimento de um protocolo, em 2007, com o Hospital Juan Canalejo (A Coruña) e com o Hospital de Santa Marta (Lisboa), os doentes com idade inferior a 65 anos foram considerados para a realização de transplante pulmonar. Uma vez que esta série inclui apenas doentes diagnosticados a partir de 2000, há ainda uma percentagem baixa de doentes transplantados. Nove doentes (7,4%) foram referenciados para a realização de transplante pulmonar, tendo sido realizado o transplante unipulmonar em 6 doentes (2 foram excluídos por infeção por Aspergillus fumigatus). O outro doente encontra-se em lista de espera.
A evolução clínica (n=76) foi, na maioria dos casos, lentamente progressiva (72,3%). Em 10 doentes (13,2%), a FPI teve uma progressão rápida e 11 (14,5%) doentes apresentaram uma EA-FPI durante a evolução da doença.
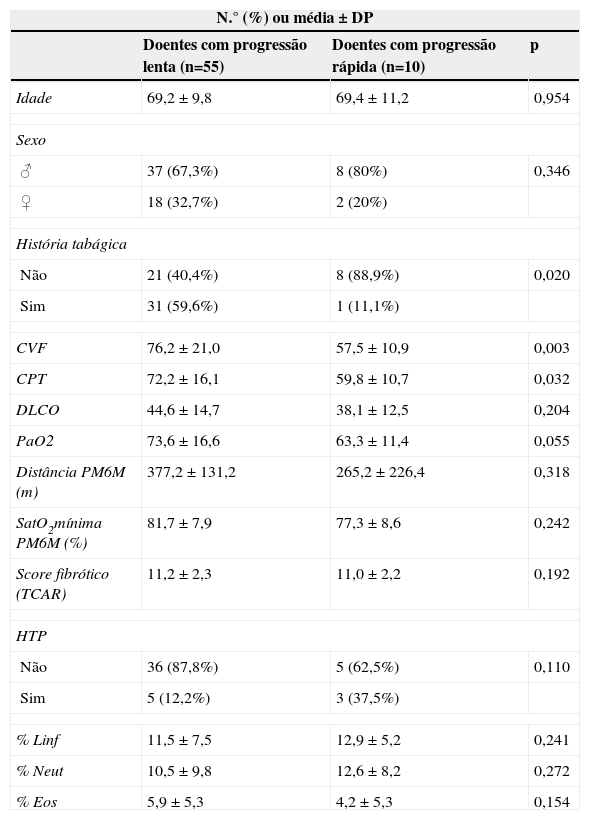
A progressão rápida da FPI (tabela 2) esteve associada à maior gravidade funcional ao diagnóstico, nomeadamente no que respeita à CVF (p=0,003) e à CPT (p=0,032). A história tabágica esteve associada a uma evolução lentamente progressiva (p=0,020).
Fatores na altura do diagnóstico e sua associação com a evolução clínica (progressão lenta versus rápida)
| N.° (%) ou média ± DP | |||
|---|---|---|---|
| Doentes com progressão lenta (n=55) | Doentes com progressão rápida (n=10) | p | |
| Idade | 69,2 ± 9,8 | 69,4 ± 11,2 | 0,954 |
| Sexo | |||
| ♂ | 37 (67,3%) | 8 (80%) | 0,346 |
| ♀ | 18 (32,7%) | 2 (20%) | |
| História tabágica | |||
| Não | 21 (40,4%) | 8 (88,9%) | 0,020 |
| Sim | 31 (59,6%) | 1 (11,1%) | |
| CVF | 76,2 ± 21,0 | 57,5 ± 10,9 | 0,003 |
| CPT | 72,2 ± 16,1 | 59,8 ± 10,7 | 0,032 |
| DLCO | 44,6 ± 14,7 | 38,1 ± 12,5 | 0,204 |
| PaO2 | 73,6 ± 16,6 | 63,3 ± 11,4 | 0,055 |
| Distância PM6M (m) | 377,2 ± 131,2 | 265,2 ± 226,4 | 0,318 |
| SatO2mínima PM6M (%) | 81,7 ± 7,9 | 77,3 ± 8,6 | 0,242 |
| Score fibrótico (TCAR) | 11,2 ± 2,3 | 11,0 ± 2,2 | 0,192 |
| HTP | |||
| Não | 36 (87,8%) | 5 (62,5%) | 0,110 |
| Sim | 5 (12,2%) | 3 (37,5%) | |
| % Linf | 11,5 ± 7,5 | 12,9 ± 5,2 | 0,241 |
| % Neut | 10,5 ± 9,8 | 12,6 ± 8,2 | 0,272 |
| % Eos | 5,9 ± 5,3 | 4,2 ± 5,3 | 0,154 |
HTP: hipertensão pulmonar; SatO2: saturação de oxigénio; % Eos: percentagem de eosinófilos na contagem diferencial do LBA; % Linf: percentagem de linfócitos na contagem diferencial do LBA; % Neut: percentagem de neutrófilos na contagem diferencial do LBA.
A morte da maioria dos doentes com FPI (69,6%) esteve relacionada diretamente com a doença – insuficiência respiratória, enquanto 13 doentes (23,2%) morreram por causas não relacionadas com a doença. Quatro doentes (7,1%) morreram por complicações pós-transplante pulmonar.
Ocorreu uma EA-FPI em 11 doentes. Entre estes, 6 (54,5%) morreram no decurso deste episódio e 2 foram submetidos a transplante pulmonar, falecendo por complicações pós-transplante.
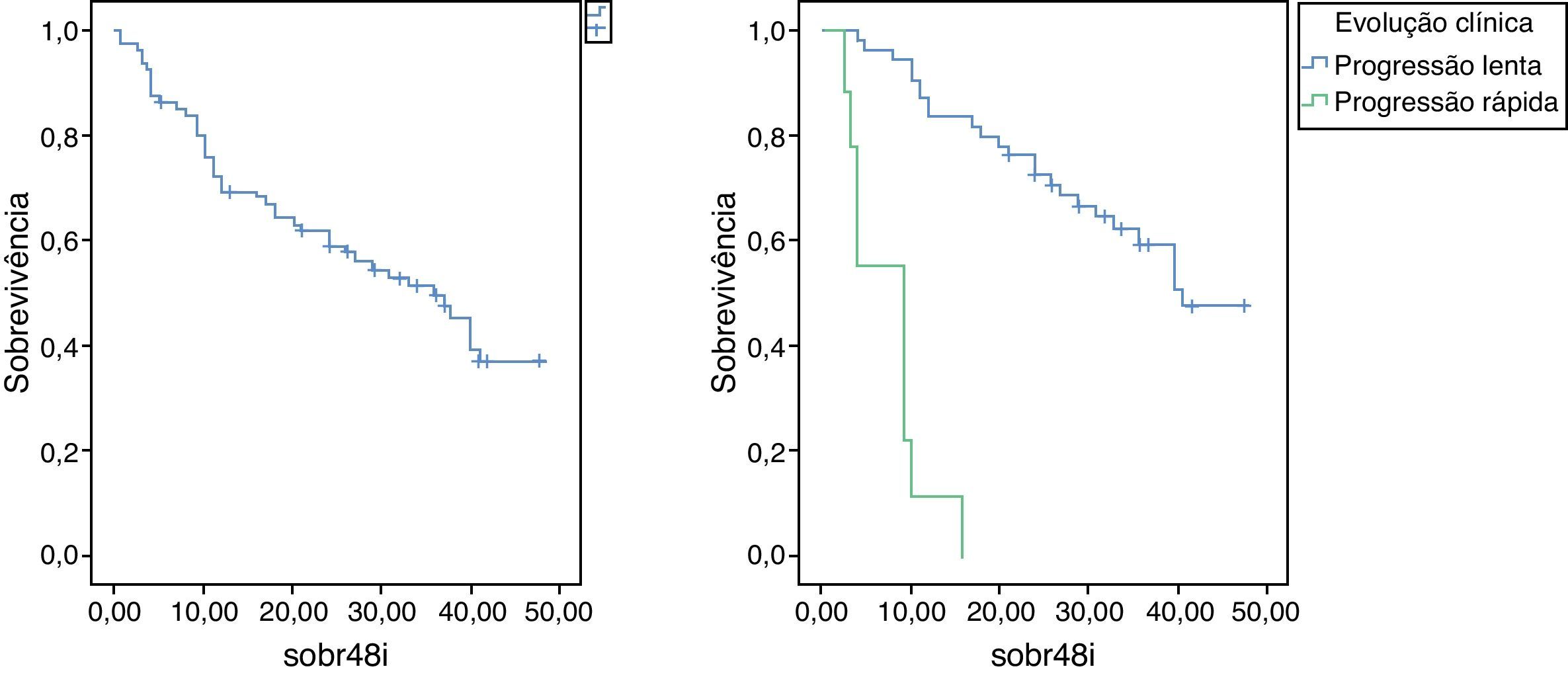
SobrevivênciaA sobrevivência mediana foi de 36 meses (fig. 1). À data de avaliação, 25 doentes permaneciam vivos.
 e para os doentes com progressão lenta e rápida (direita) – meses.")
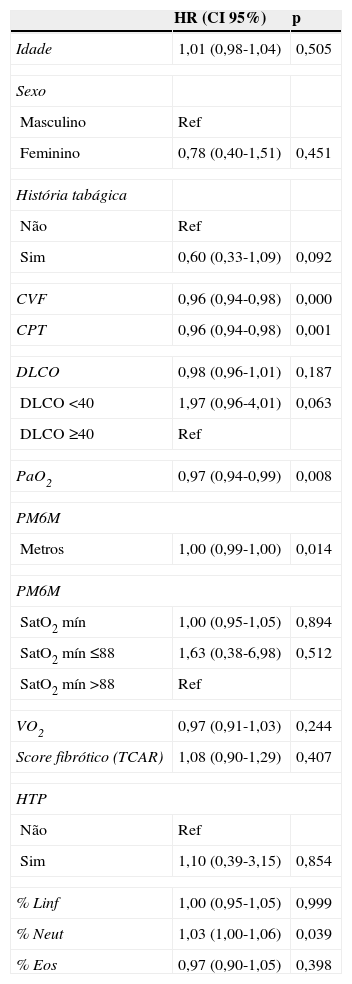
Os fatores associados à redução da sobrevivência, em análise univariada (tabela 3), foram valores inferiores de CVF (p=0,000), CPT (p=0,001), distância da PM6M (p=0,014) e PaO2 (Pressão arterial de Oxigénio) em repouso (p=0,008), bem como o aumento da contagem de neutrófilos no LBA (p=0,039).
Fatores prognósticos na sobrevivência dos doentes com FPI
| HR (CI 95%) | p | |
|---|---|---|
| Idade | 1,01 (0,98-1,04) | 0,505 |
| Sexo | ||
| Masculino | Ref | |
| Feminino | 0,78 (0,40-1,51) | 0,451 |
| História tabágica | ||
| Não | Ref | |
| Sim | 0,60 (0,33-1,09) | 0,092 |
| CVF | 0,96 (0,94-0,98) | 0,000 |
| CPT | 0,96 (0,94-0,98) | 0,001 |
| DLCO | 0,98 (0,96-1,01) | 0,187 |
| DLCO <40 | 1,97 (0,96-4,01) | 0,063 |
| DLCO ≥40 | Ref | |
| PaO2 | 0,97 (0,94-0,99) | 0,008 |
| PM6M | ||
| Metros | 1,00 (0,99-1,00) | 0,014 |
| PM6M | ||
| SatO2 mín | 1,00 (0,95-1,05) | 0,894 |
| SatO2 mín ≤88 | 1,63 (0,38-6,98) | 0,512 |
| SatO2 mín >88 | Ref | |
| VO2 | 0,97 (0,91-1,03) | 0,244 |
| Score fibrótico (TCAR) | 1,08 (0,90-1,29) | 0,407 |
| HTP | ||
| Não | Ref | |
| Sim | 1,10 (0,39-3,15) | 0,854 |
| % Linf | 1,00 (0,95-1,05) | 0,999 |
| % Neut | 1,03 (1,00-1,06) | 0,039 |
| % Eos | 0,97 (0,90-1,05) | 0,398 |
VO2: consumo de oxigénio na prova de exercício cardiopulmonar. HR: Hazard ratio
Não foi possível estabelecer pontos de corte para cada um dos fatores prognósticos encontrados, devido ao baixo poder estatístico associado à pequena dimensão da amostra.
Foram testados os pontos de corte dos fatores prognósticos basais descritos no último documento de consenso3 (DLCO <40% do valor previsto; dessaturação ≤88% durante a PM6M; presença de hipertensão pulmonar) – verificando-se que, apesar de seguirem a tendência esperada, não foram encontradas associações estatisticamente significativas.
Não foi encontrada diferença estatisticamente significativa na sobrevivência entre os doentes com FPI com ou sem enfisema.
A sobrevivência mediana foi de 41 meses nos doentes com evolução lenta e de 9 meses nos de evolução rápida (fig. 1).
Entre os doentes com evolução lentamente progressiva, valores inferiores de CVF (p=0,001), CPT (p=0,012) e distância na PM6M (p=0,026) estiveram associados à redução da sobrevivência.
Não foram encontrados fatores prognósticos nos doentes de evolução rápida (baixo poder estatístico pela reduzida dimensão da amostra).
DiscussãoA avaliação desta coorte de doentes com FPI provenientes da região norte de Portugal mostrou as características habituais associadas a esta patologia, nomeadamente no que concerne aos seus parâmetros clínicos, funcionais e radiológicos, bem como ao seu prognóstico. Nesta série, apesar da associação entre alguns fatores prognósticos à data do diagnóstico e a sobrevivência global, o principal enfoque está no tipo de evolução clínica, mais precisamente na distinção entre os doentes de evolução lenta e de evolução rápida, dado o seu prognóstico e sobrevivência serem totalmente distintos.
A idade média ao diagnóstico é consistente com o que está descrito na literatura3,24, tal como a maior prevalência no sexo masculino. A FPI familiar pode estar subvalorizada pelo facto de alguns doentes referirem morte de familiares por doença respiratória não especificada. O tabaco está fortemente associado à FPI, nomeadamente naqueles com maior carga tabágica (>20 UMA)25,26. Neste estudo, a proporção de fumadores e ex-fumadores foi inferior ao esperado, embora resultados semelhantes estejam descritos noutras séries27,28. Contudo, nos 15 doentes diagnosticados em idade inferior a 60 anos, 80% apresentava hábitos tabágicos presentes ou prévios, podendo assim especular-se que, excluindo a idade, a história tabágica pode ter sido um fator de risco para a FPI nesta amostra. O tempo mediano entre a data de início dos sintomas e o diagnóstico foi de 12 meses. Kim et al.1 descreveram um período sintomático de 6 meses a 2 anos a preceder o diagnóstico de FPI. De facto, não só os doentes tendem a atribuir inicialmente a dispneia ao envelhecimento, mas também os médicos de Cuidados de Saúde Primários tendem a focar a sua investigação etiológica em patologias mais prevalentes, tais como a patologia cardíaca e outras patologias pulmonares como a DPOC. Contudo, nesta coorte está representado um número crescente de doentes referenciados através dos Cuidados de Saúde Primários, o que poderá estar relacionado com uma maior generalização no pedido de TC torácicas.
Foi descrita recentemente uma nova síndrome, a fibrose pulmonar combinada com enfisema (CPFE), que resulta da associação entre FPI e enfisema29,30. A percentagem da CPFE está de acordo com outras publicações30. Neste estudo, foram observadas algumas características particulares desta síndrome, nomeadamente uma maior associação aos hábitos tabágicos, níveis inferiores de VEMS/CVF e um maior compromisso na capacidade de difusão. Alguns autores descrevem ainda um impacto negativo no prognóstico30, o qual está fortemente associado à ocorrência de hipertensão pulmonar30. Neste estudo, entre os doentes com CPFE e os restantes doentes com FPI, não foi encontrada uma diferença significativa na sobrevivência nem na ocorrência de hipertensão pulmonar ao diagnóstico.
Foi verificada linfocitose em 14 doentes (25%). Estes apresentaram uma linfocitose ligeira (média de 22,8%), não tendo sido verificada qualquer exposição a agentes suspeitos. É de salientar que 50% destes doentes realizaram biópsia pulmonar para excluir outros diagnósticos diferenciais, para além de não terem sido detetadas alterações clínicas, analíticas ou imagiológicas que sugerissem outras patologias associadas a UIP durante a sua evolução. Não foram igualmente encontradas diferenças na sobrevivência entre os doentes com ou sem linfocitose no LBA.
Não é possível ainda prever a evolução clínica na FPI. Nesta amostra, 13,2% dos doentes apresentou um declínio acelerado, já descrito na literatura10. Contudo, não estão ainda bem estabelecidos os critérios de classificação dos diferentes tipos de progressão. A definição para a evolução rapidamente progressiva baseia-se num tempo entre o início dos sintomas e o diagnóstico inferior a 6 meses, associado a um declínio clínico e funcional acelerado1,10. É notável a diferença na sobrevivência entre os doentes com evolução lenta e rápida (41 e 9 meses, respetivamente). Apesar de não estar ainda bem esclarecido, estes grupos poderão representar diferentes fenótipos na FPI. Em análise univariada, valores inferiores de CVF e CPT estiveram associados a uma evolução rapidamente progressiva. Os doentes não fumadores estiveram também associados a uma progressão rápida. Este facto foi já descrito noutros estudos, alguns mostrando mesmo uma melhor sobrevivência em fumadores31, não existindo para tal uma explicação clara. Alguns dados sugerem que a inalação de tabaco inibe a proliferação dos fibroblastos no pulmão, bem como a quimiotaxia, podendo assim, de alguma forma, comprometer o mecanismo de reparação patológico que se verifica após a lesão do epitélio alveolar32. Um estudo recente contradiz este facto, mostrando um melhor prognóstico nos não fumadores, e assumindo que o melhor prognóstico previamente descrito nos fumadores se deve ao facto de estes apresentarem uma doença menos grave na altura do diagnóstico, dado estarem mais alertados para eventuais sintomas respiratórios (healthy smoker lead-time effect)33.
A EA-FPI é um dos eventos mais enigmáticos e temíveis neste contexto. Os resultados deste estudo estão de acordo com a literatura publicada, quer no que respeita à incidência, quer na mortalidade. De facto, de acordo com 2 dos estudos de maior relevância34,35, a incidência da EA-FPI é de 10-15% dos doentes com FPI, e apresenta uma mortalidade elevada, entre os 50 e os 100%12,36–38. Excluindo os doentes submetidos a transplante pulmonar, a mortalidade intra-hospitalar da EA-FPI neste estudo foi de 66,7%. Têm sido procurados fatores preditores da ocorrência de EA-FPI, sem sucesso38. De facto, a ocorrência de EA-FPI não parece estar relacionada com a gravidade das alterações funcionais38. Neste estudo, nenhum dos fatores estudados conseguiu prever a sua ocorrência.
A sobrevivência mediana da amostra estudada está em linha com os outros estudos publicados39,40. Os preditores de mortalidade na FPI têm sido divididos em preditores basais (na altura do diagnóstico) e dinâmicos41. De facto, o prognóstico da FPI deve integrar parâmetros, quer basais, quer longitudinais. Neste estudo, foram apenas avaliados os preditores basais. Em análise univariada, os fatores associados à redução da sobrevivência nesta coorte estiveram relacionados com uma maior gravidade funcional (CVF, CPT), pior oxigenação (PaO2), redução da tolerância ao exercício (distância na PM6M) e maior contagem de neutrófilos no LBA.
Pela revisão da literatura, é notória a grande variabilidade de fatores prognósticos. Vários fatores contribuem para isso, nomeadamente a análise de séries com um pequeno número de doentes ou a inclusão de doentes com comorbilidades que poderão atuar como fatores confundidores. Por outro lado, a maioria dos estudos tem diferentes abordagens diagnósticas e terapêuticas no que respeita aos doentes incluídos não devendo este facto afetar o rigor dos resultados, dada a ausência de terapêutica farmacológica de eficácia comprovada. Esta coorte foi dividida de acordo com os fenótipos dos doentes, uma vez que os fatores prognósticos devem ser adaptados a cada um dos 3 tipos de evolução clínica descritos, dada a sua discrepância. Os fatores prognósticos encontrados, mais precisamente valores inferiores de CVF, CPT e distância na PM6M, permaneceram como fatores prognósticos estatisticamente significativos no grupo de doentes com evolução lentamente progressiva. O facto de não haver fatores prognósticos identificados no grupo de progressão rápida pode ser explicado pelo pequeno número de doentes, insuficiente para um resultado com significado estatístico.
Vários estudos têm descrito o valor prognóstico do estudo funcional respiratório basal, indicando que uma maior gravidade funcional está significativamente associada à redução da sobrevivência. Os fatores habitualmente descritos são a CVF27,42–44 e a CPT42,44, com maior evidência no que respeita ao primeiro. A associação da DLCO à redução da sobrevivência é um dado consistente na literatura42–45. De facto, a menor capacidade de difusão reflete uma maior extensão da fibrose e, desta forma, a maior gravidade da doença. Contudo, neste estudo, não apresentou impacto no prognóstico. Alguns estudos referem a PaO2 basal em repouso como um preditor de mortalidade8,27,43,46, embora existam também alguns resultados contraditórios44,47. Níveis mais graves de hipoxemia ocorrem em estadios mais avançados da doença, não estando ainda bem estabelecido se este pode ou não ser considerado um fator prognóstico. Vários estudos descreveram ainda o impacto prognóstico dos parâmetros da PM6M na sobrevivência48–51, quer no que respeita à distância percorrida, quer à dessaturação. Contudo, estes resultados estão provavelmente subestimados pela falta de estandardização e reprodutibilidade deste teste.
A neutrofilia no LBA tem sido associada à gravidade da doença, tal como ocorre noutras DPD como a sarcoidose52. Existe ainda evidência de que um maior grau de neutrofilia pode estar associado à redução da sobrevivência53. Embora ela esteja constantemente descrita como um fator prognóstico em análise univariada, vários estudos publicados não confirmam esta associação em análise multivariada54,55.
No contexto da FPI, a hipertensão pulmonar tem uma patogenia multifatorial, sendo, em alguns estudos, geralmente longitudinais, apontada como um preditor de mortalidade56,57. Nesta amostra, a presença de hipertensão pulmonar ao diagnóstico não teve valor prognóstico.
Foram propostos alguns coeficientes21,58 de forma a combinar diferentes parâmetros. Por exemplo, o Composite Physiologic Index, que inclui a CVF, VEMS, DLCO e a extensão da fibrose na TCAR, tem-se mostrado eficaz em vários estudos21,59. O uso destes coeficientes poderá prever com maior rigor o prognóstico e constituir uma medida mais precisa da progressão da doença.
Em conclusão, a FPI está inevitavelmente associada a um mau prognóstico. Diferentes fenótipos parecem estar a emergir, como se pode concluir das substanciais diferenças na evolução clínica. Na altura do estabelecimento do diagnóstico, uma previsão mais exata do prognóstico e evolução da doença poderia melhorar a abordagem dos doentes, particularmente no momento de referenciação para o transplante pulmonar, a única terapêutica com benefício comprovado na sobrevivência.
Conflito de interessesOs autores declaram não haver conflito de interesses.
Agradecimento especial à Dra. Carla Damas, responsável pela consulta de Transplante Pulmonar do Hospital de São João, pela estreita cooperação na referenciação dos doentes com FPI para transplante, quando indicado.


