

Job's syndrome or Hyperimmunoglobulin E syndrome (HIES) is a rare primary immunodeficiency characterized by recurrent soft tissue infections, coarse face, skeletal and vascular abnormalities, and markedly high levels of Immunoglobulin E. Eczema that resembles atopic dermatitis but is refractory to traditional treatment and severe and recurrent bacterial pneumonias often recognized during childhood. Early diagnosis and treatment prevent progressive pulmonary sequellae and increase survival. About 200 cases of HIES has been reported worldwide. The authors report a new case of HIES with one of the worst pulmonary sequellae found in the literature on this subject and review this infrequent topic.
A síndrome de Job, ou síndrome de Hiperimunoglobulina E (HIES), é uma rara imunodeficiência primária caracterizada por infecções recorrentes de tecidos moles, anomalias grosseiras faciais, esqueléticas e vasculares, e níveis visivelmente elevados de Imunoglobulina E. O eczema semelhante à dermatite atópica, mas refractário ao tratamento tradicional e pneumonias graves e recorrentes são frequentemente reconhecidas durante a infância. Um diagnóstico e tratamento precoces impedem sequelas pulmonares progressivas e aumentam a sobrevivência. Foram relatados cerca de 200 casos de HIES em todo o mundo. Relatamos um novo caso de HIES com uma das sequelas pulmonares mais extensas encontradas na análise bibliográfica sobre este assunto, conjuntamente com uma revisão sobre esta patologia pouco frequente.
The prevalence of primary immunodeficiency (PI) is underestimated worldwide. In the United States, the calculated prevalence of diagnosed PI is 1 in 1200 people of all ages.1 Hyperimmunoglobulin E syndrome (HIES) is among the earliest described syndromes of immunodeficiency characterized by recurrent and severe pneumonia, eczema, and markedly high levels of immunoglobulin E (IgE). Both male and female are affected and familial or sporadic cases have been reported worldwide. The authors present a new case of HIES, lately diagnosed, with severe pulmonary sequellae.
Case historyA 25‐year‐old man came to the hospital complaining of mild shortness of breath, cough with a yellowish expectoration, weakness and profuse unexplained sweating without fever. The first symptoms had started three month before. He was a nonsmoker and had no history of environmental exposure to chemicals or dust. His medical history was positive for dermatitis since the first 24h of his life, recurrent severe staphylococcal pneumonias with empyema, chronic external otitis, sinusitis and dermatitis. He also showed pathological fractures of the seventh left rib, intercostal herpes‐zoster at two years of age. Frequent episodes of severe infectious diarrhea were also reported. His family history was unremarkable.
On physical examination, the patient's body size was smaller than normal for his age and sex. His face was coarse, with prominent forehead, broad nasal bridge, and facial asymmetry. Marked joint hyperextensibility and scoliosis was found. Chest examination showed a respiratory rate of 24 breaths/min with crackles in the left base. His skin showed signs of pruritic chronic dermatitis spread all over, mainly at his legs, arms, and axillae. Onychomycosis was present at his fingernails with clubbed fingers. The rest of the vital signs and physical examination were unremarkable.
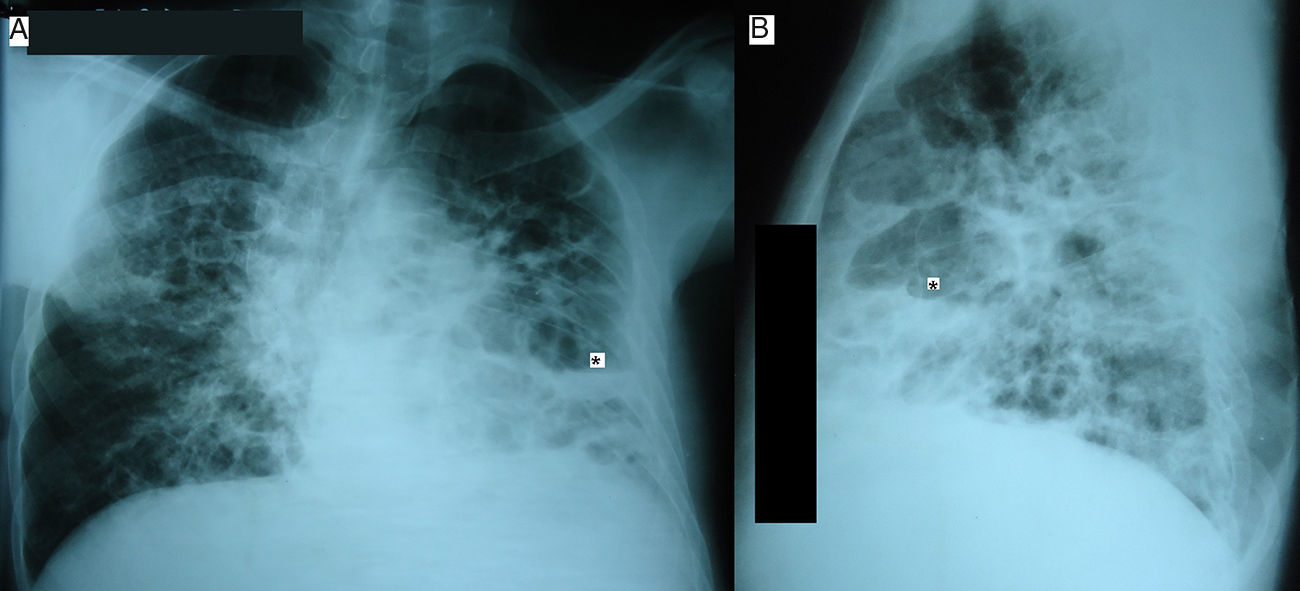
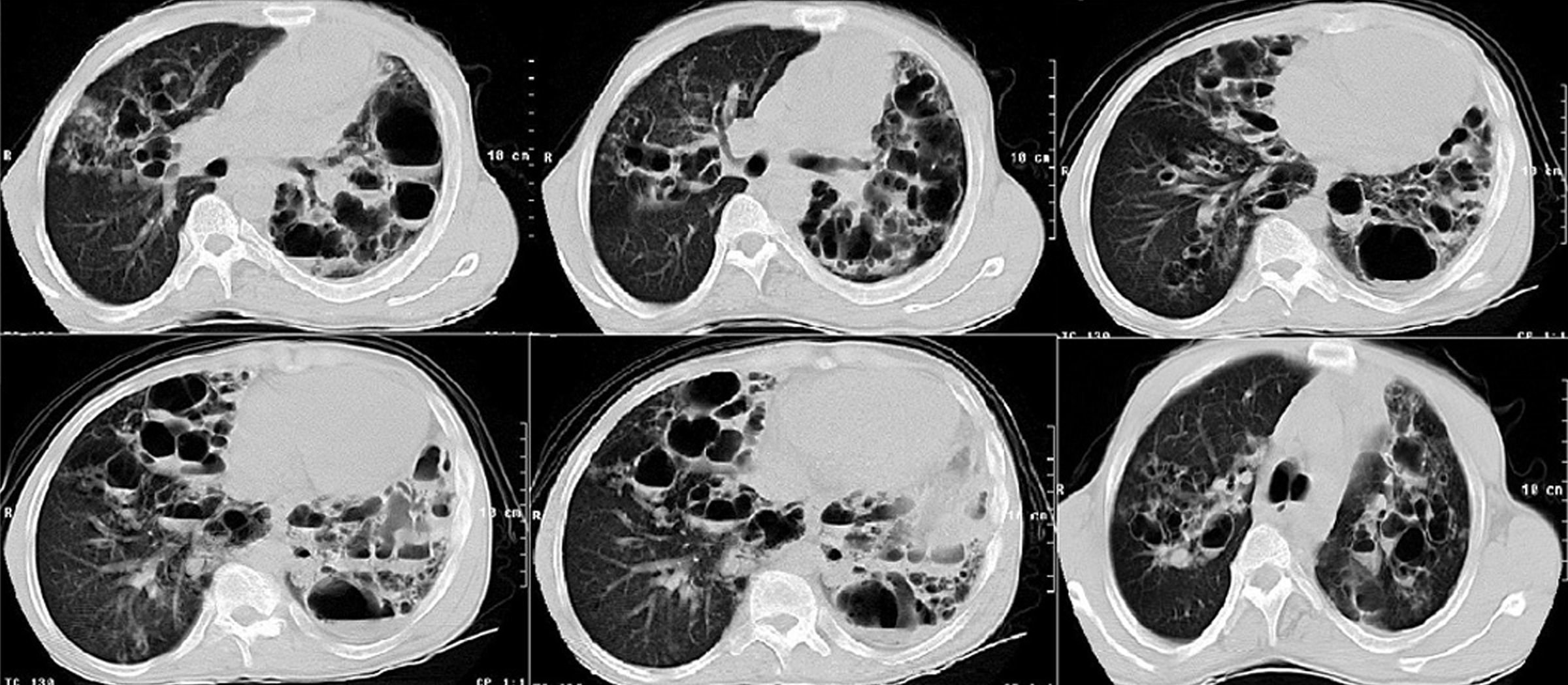
Laboratory findings included: Hb: 11.0g/dL, WBC count: 15.0×109/L with 10% eosinophils and 52% neutrophils, erythrocyte sedimentation rate: 80mm/h, alkaline phosphatase: 211U/L [reference range (RR): 35–150U/L]. No other metabolic abnormalities were observed. Alfa‐1 antitrypsin, serum IgA and IgM levels were within normal limits. IgG: 17.0g/L (RR: 6.4–13.5g/L) and IgE: 21,300U/mL (RR: 10–179U/ml) were high. A chest radiograph performed on admission showed multiple thin‐walled cysts with air‐fluid level consistent with superinfection (Fig. 1). The thorax CT scan alterations are showed in Fig. 2.
 showing multiple thin‐walled cyst (pneumatoceles) with air‐fluid level (*).")

Topical antibiotic and steroids creams, oral ketoconazole, and ceftriaxone and azithromycin were initially used without a significant improvement. On the fifth day of treatment a sputum culture with meticillin resistant S. aureus was received and vancomicin was initiated with excellent results. No other germs were isolated on blood or sputum cultures. The diagnosis of sporadic form of HIES (Job's syndrome) was made based on clinical and laboratory findings. Skin lesions improved and prophylactic treatment with trimethoprim‐sulfamethoxazole was recommended.
DiscussionThere are two forms of HIES: an autosomal‐dominant form caused by mutations in signal transducer and activator of transcription (STAT)3 (gene location 17q21),2 and a recessive form frequently associated with dedicator of cytokinesis (DOCK)8 mutations (9p24) or tyrosine kinase (TYK)2 (19p13.2) mutations. The dominant form (familial or sporadic) is characterized by skeletal, connective tissue, recurrent pulmonary infections and eczema. Autosomal‐recessive HIES is present with recurrent viral and staphylococcal skin infections, frequent central nervous system abnormalities and vasculitis. This form exhibits a higher mortality and a lack of tendency to pneumatoceles formation.3,4 Inheritance usually follows an autosomal dominant pattern with variable penetrance being the autosomal recessive pattern infrequent.
Pathogenesis is complex and not very well understood. Originally, HIES was consider to be due to a defect in neutrophil chemotaxis but this is not a constant feature. Leucocytes of HIES patients have defective cytokine‐mediated signal transduction through the Janus Kinase (JAK)‐STAT pathways. STAT3 is a major signal transduction protein involved in diverse pathways including wound healing, angiogenesis, immune pathways, and cancer. HIES patients have impaired differentiation of Th17 T cells from mutations in STAT3.5 Experimental models in which Th17 cells or Th17 cell‐dependent cytokines are defective have shown that immunity to several extracellular bacteria, including Staphylococcus aureus, Klebsiella pneumoniae and Candida albicans, is impaired.6
IgE synthesis is normally enhanced by the Th2 cytokines IL‐4 and IL‐13, and repressed by Th1 IFN‐γ and IL‐12. The two responses act in opposition to each other. Decreased production of IFN‐γ by T cells has been demonstrated in HIES patients and is probably related with hyper‐IgE.7 Eosinophilia almost always is present in these patients, at least at some point, but is not correlated with the serum IgE level or with the susceptibility to severe infections; some older patients whose IgE levels were consistently low still suffered recurrent infections.
A newborn rash is usually the first manifestation of STAT3 deficiency characterized by a fine papular erythematous eruption with superficial vesicles and pustules. This rash which may be located to the face, scalp, or body folds and is refractory to traditional topical treatment. These areas are typically spared in atopic dermatitis. Boils, cold abscesses, and chronic onychomycosis can be found.4
Severe and recurrent bacterial pneumonias are often recognized during childhood. The diagnosis is frequently delay due to the lack of evident inflammatory signs contributing to the advance of the infection and pneumatoceles and bronchiectasis formation that predispose to secondary infections. Frequent pathogens isolated in HIES are S. aureus, Streptococcus pneumoniae, H. influenzae, and Candida that typically respond promptly to appropriate antimicrobial therapy. Secondary infections are often caused by Pseudomonas aeruginosa, Aspergillus, and nontuberculous mycobacterias and have been more frequently associated with complications and mortality than primary infections.8
Characteristic craniofacial features resemble one another HIES patients more than their own family members. Facial characteristics include coarse face, prominent forehead, a broad nasal bridge, prognathism, facial asymmetry, and retained primary teeth. Skeletal manifestations such as joint hyperextensibility, scoliosis, osteopenia, pathologic fractures, and Chiari 1 malformation have been reported.4 Vascular alterations such as dilation, tortuosity and aneurysms of middle‐sized arteries producing myocardial and brain lacunar infarction, as well as arterial hypertension have been observed.9 The incidence of malignant disease in HIES is higher, particularly non‐Hodgkin's and Hodgkin's lymphoma.
Diagnosis is made based on: (1) clinical findings ‐mentioned above‐ guided by a scoring system originally developed with genetic linkage studies10: a patient with a punctuation higher than 14 is considered affected, (2) lab findings that include mild to moderate eosinophilia in over 90% of patients and markedly high IgE (>10 times normal) levels, and (3) genetic tests. The latest should be performed for confirmation but mutations are not always found and genetic analyses are not available in all medical centers. The most important differential diagnosis for HIES eczema is atopic dermatitis. Other PI disorders like Omenn syndrome, Wiskott–Aldrich syndrome, and Netherton syndrome have to be ruled out.11
Treatment of patient with HIES focuses on the following:
- 1.
Skin care. Effective skin care often depends on control of skin infections. Bleach baths and swimming in chlorinated pools are highly effective in patients with severe dermatitis. Topical corticosteroids and antibiotics, and hydrating measures for eczema, is essential.
- 2.
Antimicrobial prophylaxis. S. aureus infection prophylaxis is often given. Cotrimoxazole (2.5mg/kg of the trimethoprim component twice daily), a semisynthetic penicillin, or an oral cephalosporin which targets S. aureus can be used. Frequency and length of antibiotic prophylaxis depends on medical decision because no controlled trials have been performed. The role of antifungal prophylaxis is unknown.
- 3.
Treatment of infections. Systemic bacterial or fungal infections are often severe and a selection of the correct antibiotic drug is crucial. Intensive treatment, focusing on the most probable pathogen trying to obtain its susceptibility, could be lifesaving. Although S. aureus is the most frequent germ reported as a cause of pneumonias, gram‐negative bacteria (including Pseudomonas) and fungus has been often recognized as a cause of complications and death.8
- 4.
Management of pulmonary complications. Empyema is relatively common and requires drainage. Pulmonary large cysts may become secondarily infected and be a source of infection, bleeding, and possibly death. On the other hand, thoracic surgery can be complicated by poor expansion of the remaining lung after surgery.
- 5.
Immunomodulating agents. Intravenous immunoglobulin has been anecdotally used in HIES patients with low levels of antibodies after vaccination with inconsistent results.12 Recombinant interferon‐γ improved neutrophil chemotaxis in few patients, and vaccination for preventing infections could help HIES. However, none of these treatments are actually recommended in the daily practice because no clinical trials have been performed at the present time.11
- 6.
Hematopoietic stem cell transplantation has been used in patients with HIES and B‐cell lymphoma with negative results and is not actually indicated.13,14
- 7.
Other therapies. Isotretinoin, cyclosporin A, and plasmapheresis have been tested on a few patients but are not generally indicated.4,11 H2 antagonists like cimetidine seem to improve symptoms.
Prognosis of HIES patients depends on early diagnosis and prompt treatment of complications. Pulmonary infections, bleeding and lymphoma are among the leading causes of death. Several studies have shown that many cases of PI are diagnosed late or are never diagnosed. Some cases may be missed because the patient dies before life‐threatening complications from HIES are diagnosed.1 Identification of most children/adults with PI relies on the awareness of their physicians and a high index of suspicion leading to prompt referral to specialists trained in the management of these complex diseases.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data and that all the patients included in the study received sufficient information and gave their written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.


