

Interstitial lung disease (ILD) contributes significantly to morbidity and mortality in connective tissue disease (CTD). Early detection and accurate diagnosis are essential for informing treatment decisions and prognosis in this setting. Clear guidance on CTD-ILD screening, however, is lacking.
ObjectiveTo establish recommendations for CTD-ILD screening based on the current evidence.
MethodFollowing an extensive literature research and evaluation of articles selected for their recency and relevance to the characterization, screening, and management of CTD-ILD, an expert panel formed by six pulmonologists from the Portuguese Society of Pulmonology, six rheumatologists from the Portuguese Society of Rheumatology, and six radiologists from the Portuguese Society of Radiology and Nuclear Medicine participated in a multidisciplinary discussion to produce a joint statement on screening recommendations for ILD in CTD.
ResultsThe expert panel achieved consensus on when and how to screen for ILD in patients with systemic sclerosis, rheumatoid arthritis, mixed connective tissue disease, Sjögren syndrome, idiopathic inflammatory myopathies and systemic lupus erythematous.
ConclusionsDespite the lack of data on screening for CTD-ILD, an expert panel of pulmonologists, rheumatologists and radiologists agreed on a series of screening recommendations to support decision-making and enable early diagnosis of ILD to ultimately improve outcomes and prognosis in patients with CTD.
Interstitial lung diseases (ILDs) are a heterogeneous group of diffuse lung parenchymal disorders. They are one of the most common pulmonary manifestations of systemic rheumatic diseases that include systemic sclerosis (SSc), rheumatoid arthritis (RA), Sjögren syndrome (SS), mixed connective tissue disease (MCTD), idiopathic inflammatory myopathies (IIMs), and systemic lupus erythematosus (SLE).1–8
The epidemiology of ILD in the context of connective tissue disease (CTD) is difficult to determine due to undefined classification criteria, overlap syndromes, and undifferentiated CTD.5 Nearly 20% of ILDs reported in Europe and the USA are associated with autoimmune rheumatic diseases9 and contribute to significant morbidity and early mortality in this setting.1–6,8
Connective tissue disease-associated ILD has a highly variable presentation and clinical course, ranging from subclinical abnormalities to respiratory failure and death, even in patients with the same rheumatic disease; in addition, irreversible loss of lung function can be silent and significant in early stages.2,3,10,11 Interstitial lung diseases may precede or follow the onset of rheumatic symptoms, as pulmonary manifestations can be the first sign of rheumatic disease or occur later during disease progression. Although autoimmune-mediated lung injury is thought to have a common mechanism in CTD, its prevalence varies according to the underlying disease. It occurs, for example, in nearly 50% of patients with SSc but in just 2% to 10% of those with SLE.3,5
Early detection of pulmonary involvement and accurate ILD diagnosis are essential for informed treatment decisions and prognosis.6,8,12 Clear evidence and guidelines on screening for ILD in patients with CTD, however, are lacking.12
To define best practices in ILD screening in the context of autoimmune rheumatic diseases in Portugal, experts from the Portuguese Society of Pulmonology, the Portuguese Society of Rheumatology and the Portuguese Society of Radiology and Nuclear Medicine were invited to join a multidisciplinary panel to achieve consensus on which patients should be screened and how and on how often patients with SSc, RA, SS, MCTD, IIMs, and SLE should be screened and monitored. The aim of this article is to summarize the recommendations agreed on by the expert panel.
MethodologyIn November 2022, a narrative non-systematic literature search of MEDLINE/Pubmed database was conducted using the keywords "interstitial lung disease", “CTD-ILD”, "rheumatic diseases", "systemic sclerosis", "rheumatoid arthritis", "mixed connective tissue disease", "Sjögren syndrome", "idiopathic inflammatory myopathies" and "systemic lupus erythematous". The authors also proposed other publications throughout the preparation of the present document, which were added to the final references. Only publications with at least the abstract and in English, Portuguese or Spanish were considered. Papers concerning COVID-19 were excluded.
An expert panel, formed by six pulmonologists, six rheumatologists, and six radiologists, selected the articles for their relevance to the characterization and screening of ILDs in systemic rheumatic diseases, with a focus on recent publications.
Considering the GRADE (Grading, Recommendation, Assessment, Development and Evaluation) approach,13 the authors rated the recommendations as strong or low evidence based on the quality of evidence, desirable and undesirable effects, patient values and preferences, resources required, health equity, acceptability of the tests by key stakeholders, and feasibility of implementation of the tests. All recommendations were evaluated as having low evidence.
Interstitial lung disease assessmentInterstitial lung disease diagnosis is based on a multidisciplinary assessment of clinical, laboratory, imaging, functional, and sometimes histologic features.14,15 Initial evaluation of all patients with suspected ILD should include a complete history and physical examination. The clinical evaluation must include a search for clinical features suggestive of CTD.6,8 The presence of ‘velcro’ crackles in lung auscultation suggests the presence of lung fibrosis and should be addressed and considered if present. In the case of interstitial pneumonia, an autoimmune panel should be performed to investigate a hypothetical underlying CTD. High-resolution computed tomography (HRCT) is the primary imaging tool used to detect ILD and outline its extension and pattern.5,6,16,17 A volumetric HRCT scan should be acquired on full inspiration (slice thickness, ≤1.5 mm), complemented at baseline with an additional acquisition in ventral decubitus and a non-contiguous acquisition on expiration (slice thickness, 1 mm; 20-mm interval). In CTD, bronchoalveolar lavage and lung biopsy are generally reserved for cases where the first diagnostic impression is inconclusive18 or when an infection or lung toxicity is suspected.
Multiple ILD patterns may be observed in SSc, RA, SS, MCTD, IIMs, and SLE, although the prevalence varies according to the disease in question.8 Imaging patterns in most patients with ILD characteristically range from ground-glass opacities to honeycombing associated with volume loss.6,12 Nonspecific interstitial pneumonia (NSIP) is the main pattern observed in SSc, IIMs, and SS.19 It appears to be more common than usual interstitial pneumonia (UIP) in SLE, whereas in RA-ILD, UIP seems to be predominant.
Pulmonary function tests (PFTs), which are non-invasive, safe, cost-effective, and clinically feasible, are also a reliable tool for early ILD detection.12,20 Although PFTs do not have a role in the differential diagnosis of ILD, detection of decreased diffusing capacity for carbon monoxide (DLCO) can aid an early diagnosis. Pulmonary function tests provide the most accurate measurement of disease severity, and serial PFTs should be performed as part of a close-follow-up strategy.17
Patients diagnosed with ILD should undergo a multidisciplinary evaluation of disease severity, associated symptoms, pulmonary function impairment, and disease extent on HRCT.
Systemic sclerosisSystemic sclerosis is a rare, complex, multisystemic disease characterized by immunological events, vasculopathy, and cellular inflammation that cause excessive fibrosis of the skin and multiple internal organs.2,4,21 The extent and distribution of skin fibrosis determine the phenotype of SSc, which can be classified as diffuse cutaneous (dcSSc) or limited cutaneous (lcSSc)2,4,21
Interstitial lung disease is the prevailing pulmonary manifestation of SSc; it is detected in approximately 50% of patients and is the leading cause of hospitalization, morbidity, and mortality.4,17,22–27 In an analysis of 3656 patients with SSc from the EULAR Scleroderma Trials and Research (EUSTAR) cohort, 53% of patients with dcSSc and 35% of those with lcSSc had ILD.28 Earlier studies reported ILD in up to 90% of patients with SSc.2,3 SSc-ILD risk factors include dcSSc, African-American ethnicity, male sex, older age at disease onset, smoking history, short disease duration, esophageal dysmotility/dilatation/gastroesophageal reflux, a high modified Rodnan skin score, elevated C-reactive protein (CRP), hypothyroidism, reduced forced vital capacity at diagnosis, worsening PFTs and 6-minute walk distance (6MWD) performance, and presence of anti-Th/To ribonucleoprotein and anti-topoisomerase I antibodies.4,8,17,21,29,30 Anti-centromere antibodies are considered protective and decrease the likelihood of ILD development in SSc.4,17,21,29
The course of SSc-ILD is heterogeneous. While it frequently follows a chronic, indolent course, it can develop into a progressive, even life-threatening, disease. The most frequent respiratory symptoms, such as cough and exertional dyspnea, may be absent or mild in early forms of the disease.12,31
Median survival among patients with SSc-ILD is 5 to 8 years.4 There is no clear evidence of an association between the different fibrosis patterns on HRCT and SSc prognosis or mortality, although some reports suggest a worse prognosis associated with UIP pattern.21,32,33
Systemic sclerosis-associated ILD screening and monitoringThe variable nature of SSc-ILD and lack of robust prognostic markers makes it difficult to predict which patients will develop clinically meaningful disease; there is also significant variability in risk for disease progression.5,12 All patients with SSc should undergo HRCT screening for ILD.12,17,27,31,34 Pulmonary function tests, including spirometry and DLCO, should also be performed to provide baseline parameters. Although PFTs can suggest the presence of ILD, they should not be the only primary screening tool, as lung volumes and diffusion capacity can be within normal ranges in early ILD.27 Additionally, pulmonary hypertension (PH) is more common in SSc in comparison with other CTDs and as so, patients should be screened annually with echocardiography, especially in case of a lower isolated DLCO. Regarding PH in patients with CTDs and SSc in particular, a thorough investigation should be performed to ensure accurate characterization of the PH group classification and appropriate referral.35 The functional assessment may also include the 6MWD test, which is widely recognized, simple, non-invasive, low-cost, and reproducible.12
As the risk of ILD is greatest in the first 5 years of SSc, patients should be monitored closely during this period and be referred for HRCT and/or PFTs according to individualized risk stratification.21,27,34 High-resolution computed tomography should also be considered on detection of new respiratory symptoms, physical exam alterations, PFT impairment, or oxygen desaturation.12,31
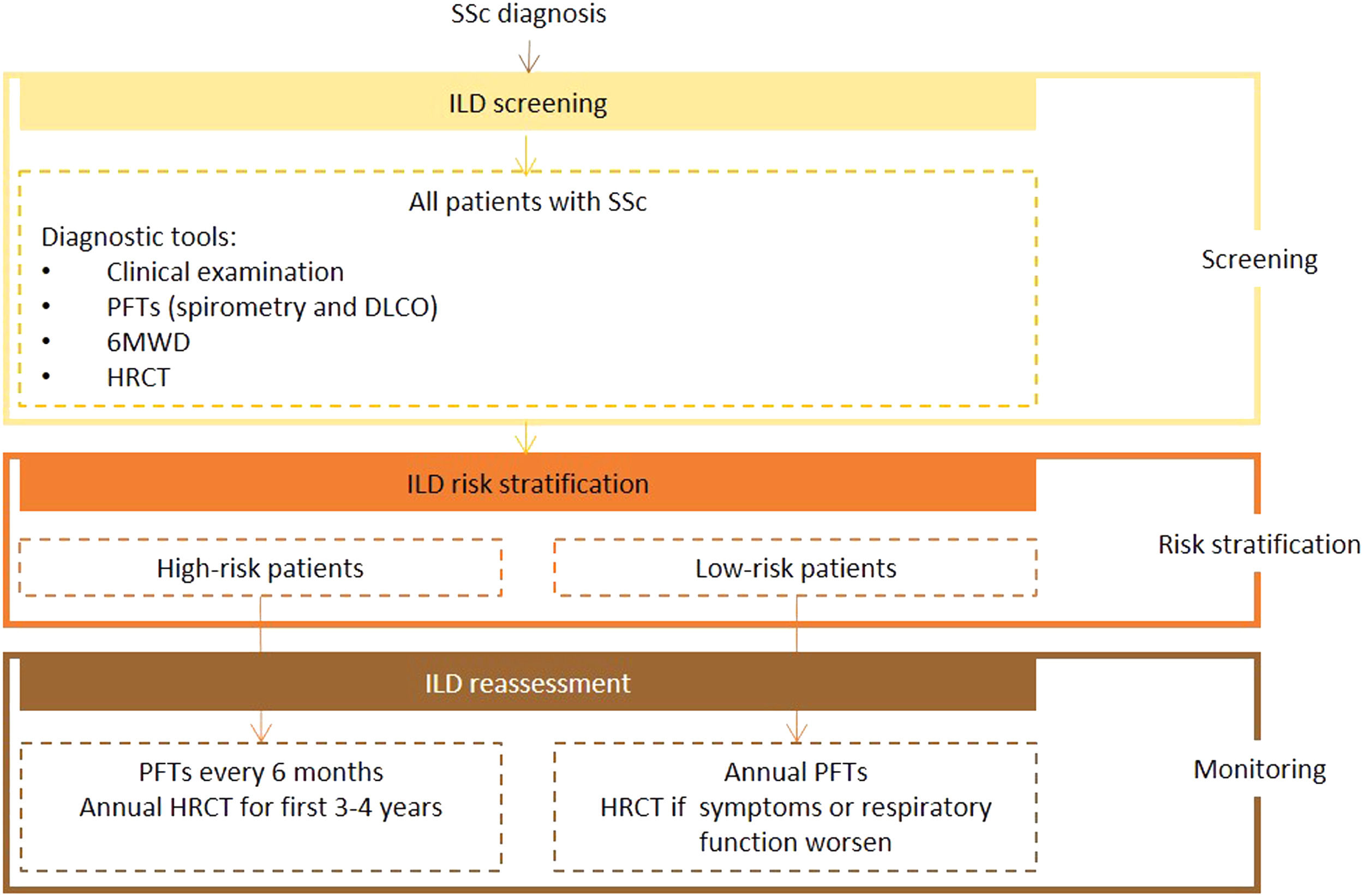
Expert panel recommendationsAll patients with SSc should be screened for ILD on diagnosis using HRTC, PFTs, and the 6MWD with auricular sensor. After this initial assessment, they should be risk stratified according to recognized predictors of ILD.
Pulmonary function tests, including spirometry and DLCO, should be performed at least every 6 months in higher-risk patients and ever year in other cases. High-resolution computed tomography should be performed annually in the first 3 to 4 years in high-risk patients. Any patient who shows worsening clinical or respiratory function, regardless of risk stratification, should be reassessed by HRCT.
Ultrasound is not recommended for diagnostic or screening purposes as it is strongly operator- and training-dependent, poorly reproducible, and supported by limited data.
Fig. 1 presents the recommended screening algorithm for ILD in patients with SSc.

Proposed algorithm for ILD diagnosis at baseline and during follow-up with respective tools and repeat screening strategy for patients with SSc. SSc - systemic sclerosis; DLCO - diffusing capacity for carbon monoxide; HRCT – high-resolution computed tomography; PFTs – pulmonary function tests; 6MWD – 6-minute walk distance test.
Rheumatoid arthritis is a chronic inflammatory rheumatic disease involving the synovial joints. Typically, symmetrical arthritis leads to cartilage and bone erosion, causing joint destruction.36 Rheumatoid arthritis may have several extra-articular manifestations, including pulmonary involvement, which is a significant contributor to morbidity and mortality and accounts for approximately 10% to 20% of related deaths.2,3,6,37
Interstitial lung disease is the most common pulmonary presentation, with an estimated prevalence of 10% to 60%.3,6,38 It can precede joint involvement in up to 20% of patients.2 In contrast to other rheumatic diseases, the most frequent subtype of RA-ILD is UIP, which is present in almost 50% of patients and is associated with a worse prognosis.3,6,39 Clinical presentation can range from asymptomatic to rapidly progressive disease. Symptoms usually progress over time, with progression rates varying according to histopathologic patterns of ILD and other clinical characteristics, such as disease extent and rate of pulmonary function decline.3,40 Clinically significant restrictive lung disease occurs in 8% to 15% of patients. Since approximately 30% of patients have subclinical ILD, HRCT should be performed in all patients with RA and respiratory symptoms, risk factors for ILD, or minor chest radiographic abnormalities, as it could lead to an earlier diagnosis.6,25,26,41
Risk factors for RA-ILD include male sex, age >55 years, longer disease duration, and higher joint disease activity, current or ever smoking, prednisone use, anti-cyclic citrullinated peptide antibodies, elevated erythrocyte sedimentation rate (ESR), rheumatoid factor positivity, a family history of RA, and presence of rheumatoid nodules or other extra-articular manifestations.3,4,6,38,42
The hazard ratio of death for patients with RA is three times higher when ILD is present.3,43 Five-year mortality rates following a diagnosis of ILD range from 35% to 39%.3,43,44 Estimated 5-year survival rates are 36% in patients with RA-UIP and 94% in those with RA-NSIP.6
Rheumatoid arthritis-associated ILD screening and monitoringInitial evaluation of patients with suspected RA-ILD should include HRCT and PFTs (spirometry and DLCO).37,45 Although universal HRCT screening for ILD in this setting seems challenging given the high prevalence of RA, limiting this test to symptomatic patients would leave a significant proportion of cases undiagnosed. Therefore, any patient with RA with risk factors for ILD or who develops respiratory symptoms, impaired lung function, or crackles on chest auscultation should be referred for HRCT.14,46 Although some authors suggest performing HRCT only in patients with certain risk factors or scores, data are lacking on the impact of individual or combined risks. In our opinion, all these factors should be considered.45 Several biological and genetic markers associated with a higher risk of ILD have been recently described, including anti-carbamylated protein antibodies, extracellular matrix metalloproteinase 7, MUC5B mutations, and mutations in telomerase genes that cause accelerated telomere shortening.46–53 The use of these markers, however, is currently restricted to research purposes. There is insufficient evidence to recommend their use in routine clinical practice.
Pulmonary function tests should be repeated regularly and, together with risk factors, used to guide the frequency of HRCT.14
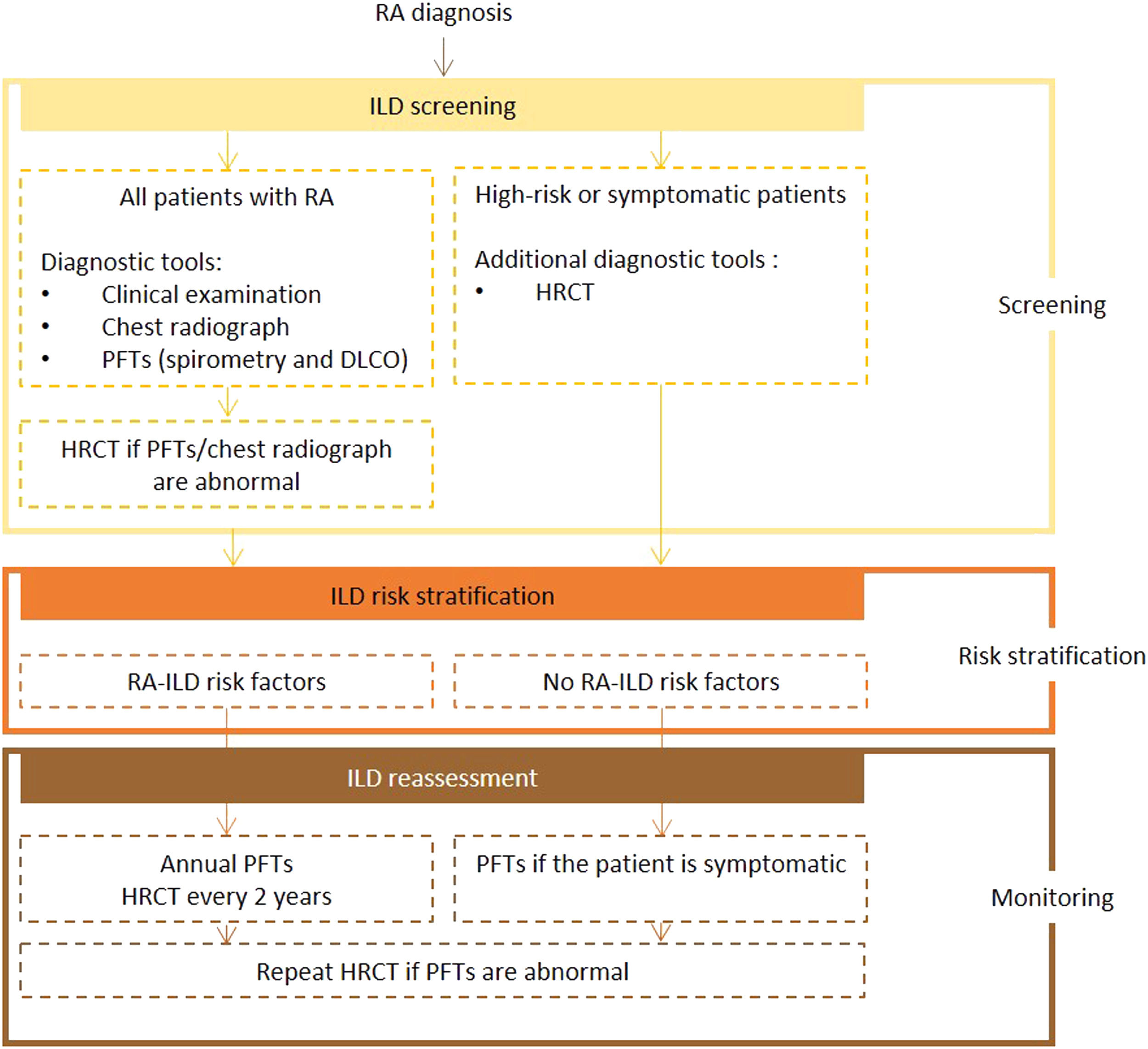
Expert panel recommendationsAll patients diagnosed with RA should be screened for ILD using PFTs (including spirometry and DLCO) and chest radiography. If abnormalities are detected, an HRCT scan should be performed. Symptomatic and at-risk patients should also undergo HRCT.
Frequency of reassessment is dictated by the presence or absence of risk factors. Screening with PFTs every year and HRCT every 2 years is recommended for high-risk patients. In low-risk patients, PFTs should be repeated if symptoms develop. If the test results are abnormal, a repeat HRCT scan is recommended.
Fig. 2 presents the recommended screening algorithm for ILD in patients with RA.

Proposed algorithm for ILD diagnosis at baseline and during follow-up with respective tools and repeat screening strategy for patients with RA. RA – rheumatoid arthritis; DLCO - diffusing capacity for carbon monoxide; HRCT – high-resolution computed tomography; ILD - interstitial lung disease; PFTs – pulmonary function tests.
Mixed connective tissue disease is a systemic rheumatic disease characterized by the combination of clinical manifestations of SSc, SLE, RA, and IIMs and positive anti-ribonucleoprotein antibodies.2,4,54–56 There is some discussion about whether MCTD is an independent entity or an early stage of one of the above CTDs or overlap syndrome.2,4,54,55,57–59
The most common clinical features of MCTD include polyarthritis, Raynaud phenomenon, puffy fingers, myositis, ILD, and esophageal dysmotility.56,60,61 Although little has been published on the prevalence, course, and outcome of MCTD-ILD,55,56,62 ILD has a significant impact on survival in this setting and should be regarded as a major complication.55,61,62 The most common radiological pattern is NSIP.2,4,6,56
Interstitial lung disease is estimated to be present in 40% to 80% of patients, but treatment during the acute inflammatory phase is associated with a good prognosis.2,4,60–63 Although ILD in MCTD is typically modest in extent, nearly 50% of patients will experience progression, which is generally slow but seems to continue for several years after diagnosis, leading to deteriorating lung function.55 Signs of fibrosis on HRCT are associated with increased mortality,60 with one study showing a mortality rate of 20.8% in patients with severe pulmonary fibrosis compared with just 3.3% in those with a normal HRCT scan.63
Mixed connective tissue disease-associated ILD risk factors include esophageal dilatation and motor dysfunction, dysphagia, Raynaud phenomenon, anti-Smith or anti-Ro-52 antibodies, rheumatoid factor, and no history of arthritis.2,55,56,59,60 High anti-ribonucleoprotein antibody titers at baseline are a strong predictor of ILD progression.55 Disease extent on HRCT also has prognostic value.62
Because of the nature of MCTD, close monitoring and prognostic stratification based on different clinical and serological features are essential for improving survival outcomes and quality of care.60
Mixed connective tissue disease-associated ILD screening and monitoringCurrently, there are no guidelines on ILD screening in patients with MCTD. Since chest radiography will not detect mild disease, and waiting for symptoms and/or a decline in lung function may be detrimental, patients should undergo HRCT to assess pulmonary involvement.62
All patients diagnosed with MCTD should undergo thoracic HRCT and PFTs (spirometry and DLCO) to check for ILD and establish baseline lung function.40 Promising results have been reported for immunological studies of immune complexes and total hemolytic complement levels (CH50/ml U) and for (99 m)Tc-DTPA aerosol clearance times, but the data are insufficient to recommend their standard use.61
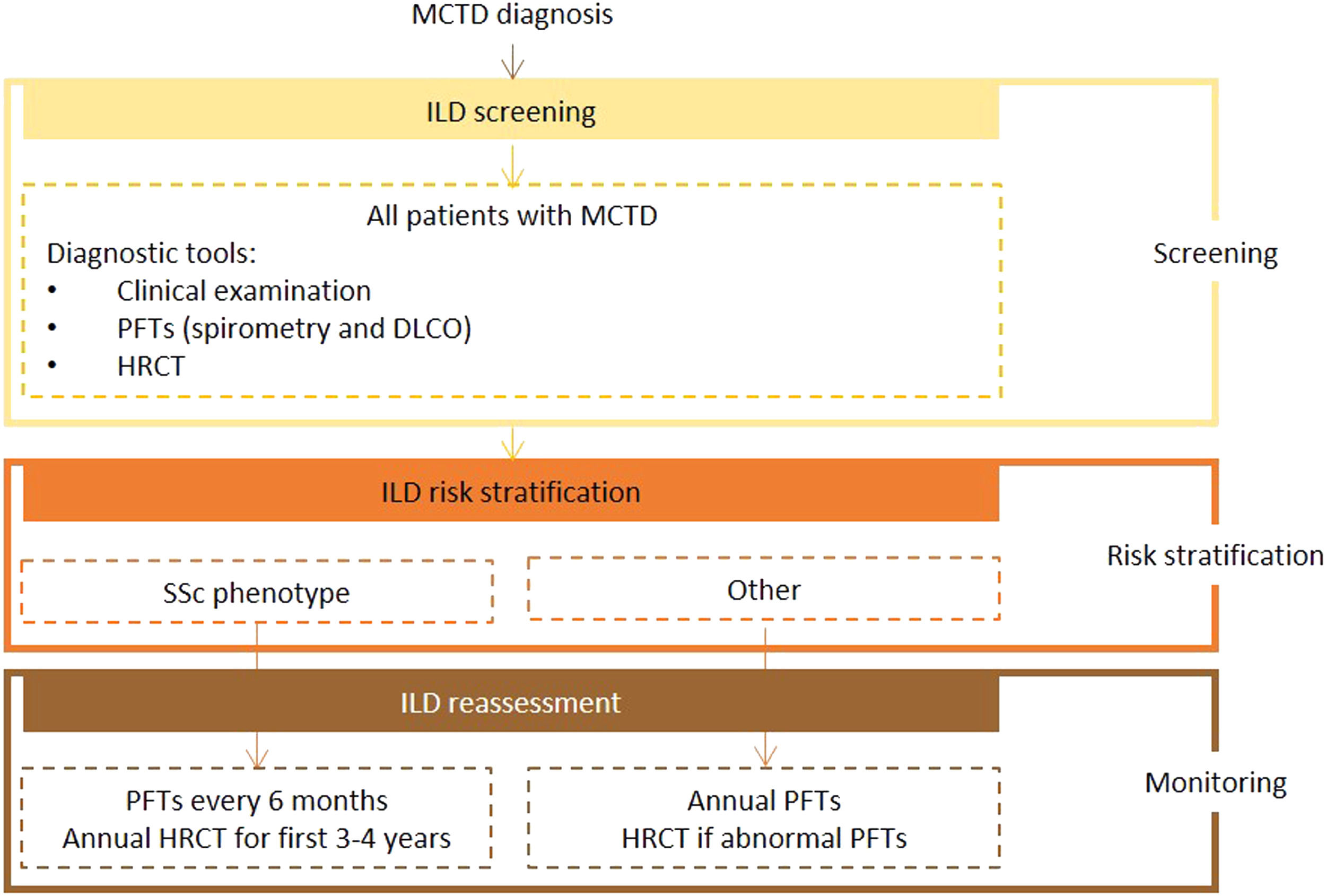
Expert panel recommendationsAlthough there was an overall lack of consensus on ILD screening recommendations for MCTD due to its heterogeneous manifestations, the expert panel agreed that all patients should undergo screening with HRCT and PFTs (spirometry and DLCO) on diagnosis. The level of evidence for this recommendation is very low because of the lack of data.
Frequency of reassessment will depend on disease course. In patients with an SSc phenotype, patients should be reassessed at similar intervals to those with SSc (PFTs every 6 months and HRCT every year for the first 3 to 4 years after diagnosis). All other patients should undergo annual clinical examination and PFTs, with referral for HRCT if abnormalities are detected.
Fig. 3 presents the recommended screening algorithm for ILD in patients with MCTD.

Proposed algorithm for ILD diagnosis at baseline and during follow-up with respective tools and repeat screening strategy for patients with MCTD. MCTD - mixed connective tissue disease; DLCO - diffusing capacity for carbon monoxide; HRCT – high resolution computed tomography; ILD - interstitial lung disease; PFTs – pulmonary function tests; SSc - systemic sclerosis.
Sjögren syndrome is a chronic systemic inflammatory disease that mainly affects the exocrine glands (in particular the lacrimal and salivary glands) through focal lymphocytic infiltration; there may also be extraglandular organ involvement affecting the joints, lung, kidneys, small vasculature, and other endocrine glands.2,4,64–66
Pulmonary involvement has been reported in between 9% and 75% of patients, with rates varying according to methods and patient selection.4,67–69 Interstitial lung disease occurs in 9% to 30% of patients with SS2,6,70 and is associated with increased morbidity, including life-threatening complications.66,68,71,72 Primary SS-ILD is associated with a worse prognosis than SS without pulmonary manifestations, with a fourfold increased risk of 10-year mortality and a reported 5-year survival rate of 85%.6,67,69,73–75
Nonspecific interstitial pneumonia is the most frequent CT pattern in SS-ILD; other patterns include UIP, lymphoid interstitial pneumonia, and organizing pneumonia.2,4,6,64,66,71,72,74–77
Sjögren syndrome-associated ILD risk factors include male sex, age ≥65 years, smoking history, a high antinuclear antibody titer, a positive rheumatoid factor test, anti-SSB/La, and/or anti-Ro52 antibodies, digestive (particularly esophageal) involvement, elevated C-reactive protein (CRP), systemic manifestations, high ESR, and hypergammaglobulinemia.4,59,64,65,67,74
Sjögren syndrome-associated ILD screening and monitoringAt the initial visit, baseline PFTs (including spirometry and DLCO) and chest radiography are recommended for patients with SS.66,72 If lung involvement is suspected, an HRCT scan may be preferable due to its higher sensitivity and specificity.78,79 It can also determine the presence and extent of ILD and inform treatment decisions.72,78
In general, all patients with SS should undergo serial monitoring with clinical, functional, and imaging evaluation. The frequency of these tests should be based on clinical judgement and the presence of risk factors for ILD.66,79 For a better understanding of longitudinal disease course, PFTs should be repeated at least every 12 months.72,79
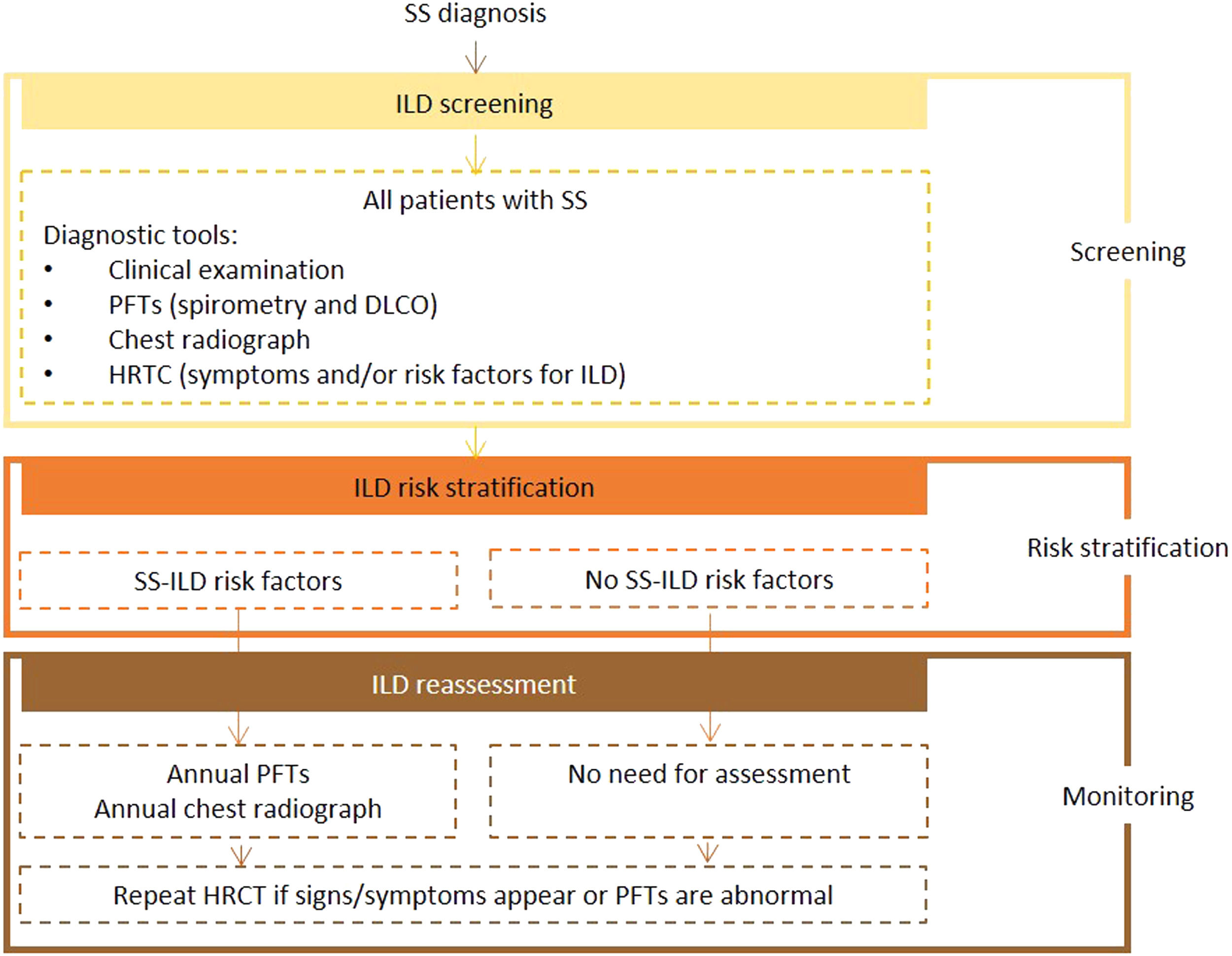
Expert panel recommendationsBaseline PFTs, including spirometry and DLCO, and chest radiography should be performed in all patients with SS. Patients with manifestations and/or risk factors for ILD should also undergo HRCT screening. The initial tests should be repeated annually in patients with risk factors, and HRCT performed in the presence of signs or symptoms.
Fig. 4 presents the recommended screening algorithm for ILD in patients with SS.

Proposed algorithm for ILD diagnosis at baseline and during follow-up with respective tools and repeat screening strategy for patients with SS. SS - Sjögren syndrome; DLCO - diffusing capacity for carbon monoxide; HRCT – high resolution computed tomography; ILD - interstitial lung disease; PFTs – pulmonary function tests.
Dermatomyositis and polymyositis are two forms of idiopathic inflammatory myopathies (IIMs), also known as myositis. Myositis is a group of rare, heterogeneous CTDs marked by skeletal muscle inflammation and frequent extramuscular involvement, often associated with lung involvement.6,80 Most myositis-associated lung involvement takes the form of ILD, with prevalence rates varying from 20% to 86%.2,3,6,81–84 As in other rheumatic diseases, ILD can appear before, after, or simultaneously with the onset of skin or muscle manifestations, but tends to be a component of early myositis.3,6,82,85,86
Myositis-associated ILD has a variable clinical course. Depending on the respiratory symptoms at presentation, it can be categorized as: 1) rapidly progressive, the most severe form, with acute onset and respiratory failure occurring within weeks; 2) chronic with slowly progressive symptoms, the most common variant (50%), presenting with insidious onset dyspnea, non-productive cough, and rarely, constitutional symptoms; 3) asymptomatic/subclinical.3,82 Up to 30% of patients with polymyositis or dermatomyositis have subclinical or asymptomatic ILD, highlighting the need for pulmonary screening in patients with myositis and any signs or symptoms of pulmonary involvement and/ or presence of IMM-ILD risk factors3,82,87 Chronic or subclinical ILD can progress rapidly during the later stages of disease and is often accompanied by fever and malaise.3,82
Although myositis-associated ILD tends to be associated with more than one imaging pattern, NSIP is the predominant pattern, followed by UIP.2,3,6,81,88,89 Treatment response also varies depending on the underlying pattern.3,90,91
The risk factors for IIM-ILD include age >45 years, presence of arthritis/arthralgia, elevated ESR and CRP, and presence of myositis antibodies, especially anti-MDA5 and the anti-aminoacyl-tRNA synthetase antibodies, which can also influence clinical presentation and disease course.2,6,8,86,91–94
Overall 5-year survival rates in patients with myositis-associated ILD range from 60% to 80%.3,6,82,95 Nonspecific interstitial pneumonia is associated with better survival than UIP.2,84,96,97
Idiopathic inflammatory myopathies-associated ILD screening and monitoringEstablishing a diagnosis of IIM-ILD, particularly in patients with rapidly progressive disease, often requires a multidisciplinary approach with comprehensive history taking, organ system review, physical examination, PFTs, and chest imaging. In addition to clinical phenotyping, evaluation of a patient with myositis should include serologic autoantibodies, which can inform treatment decisions and prognosis for patients with ILD.80,93
Guidelines on the recommended frequency of PFTs with respiratory pressures evaluation and HRCT are currently lacking for patients with IIM-ILD. Repeating these tests should be at the clinician's discretion based on the detection of worsening or new-onset pulmonary symptoms, such as cough, dyspnea, and decreased functional status.80,93
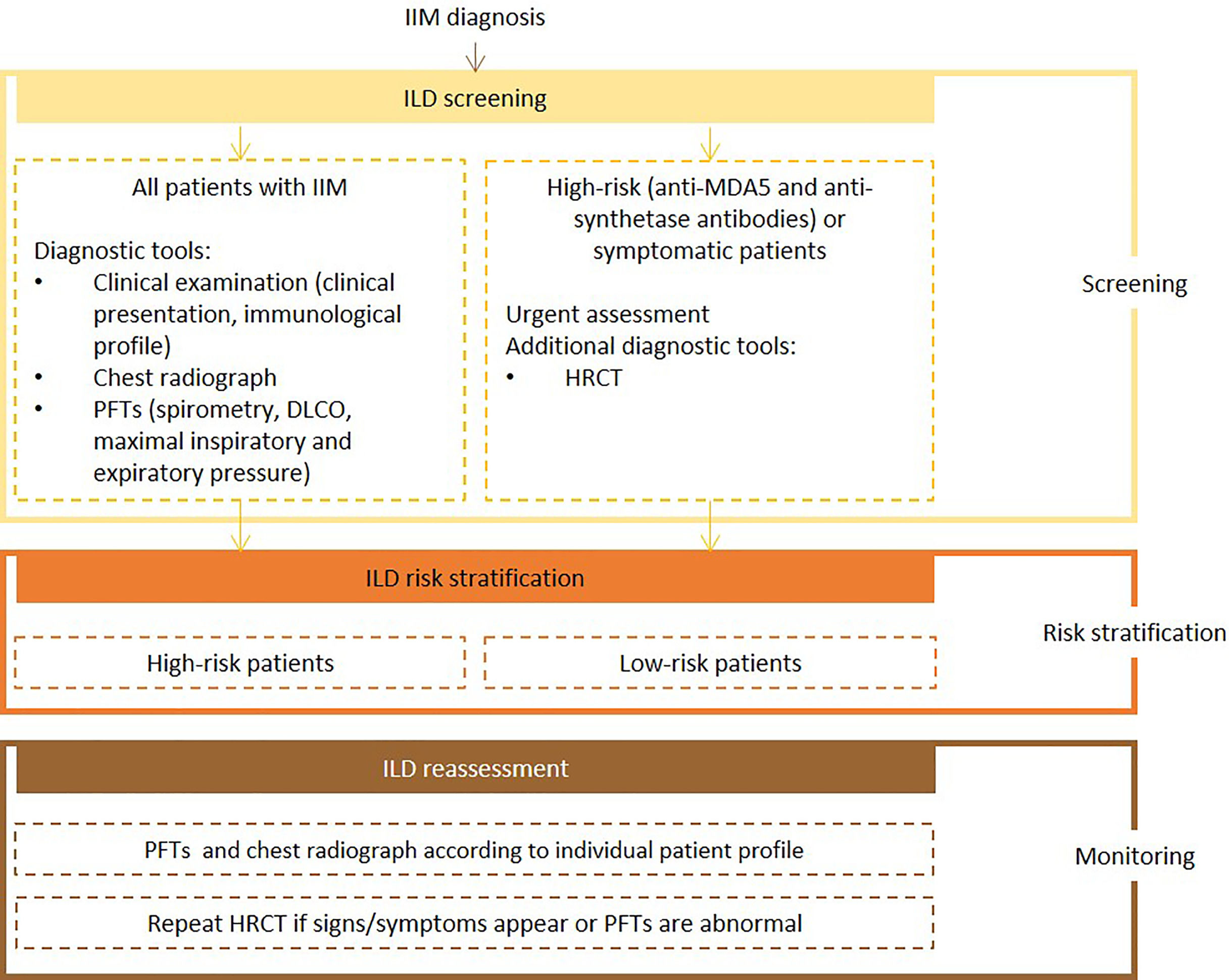
Expert panel recommendationsClinical presentation, risk factors for IIM-ILD, autoantibody profile (e.g., anti-MDA5 and anti-synthetase antibodies), chest radiograh, PFTs with spirometry, DLCO, and respiratory pressures evaluation, should be assessed at baseline for all patients with IIM. Urgent assessment is necessary for patients with anti-MDA5 and anti-synthetase antibodies which, at baseline, should also be referred for HRCT.
Frequency of reassessment will depend on the evaluation of individual patient's immunological profile, the presence of risk factors for poor prognosis and predictive factors for rapidly progressive-ILD. Pulmonary function tests and chest radiography should be repeated at the clinician's discretion. If symptoms develop, or if abnormalities are detected, HRCT should be ordered.
Fig. 5 presents the recommended screening algorithm for ILD in patients with IIM.

Proposed algorithm for ILD diagnosis at baseline and during follow-up with respective tools and repeat screening strategy for patients with IIM. IIM - idiopathic inflammatory myopathies; DLCO - diffusing capacity for carbon monoxide; HRCT – high resolution computed tomography; ILD - interstitial lung disease; PFTs – pulmonary function tests.
Systemic lupus erythematosus is a multisystem autoimmune disease with cutaneous, musculoskeletal, renal, neurologic, hematological, and pulmonary manifestations, mostly affecting women of reproductive age.2,59,98 Systemic lupus erythematosus-related pulmonary manifestations can affect any component of the lung structure,3 are wide-ranging and debilitating in nature, and usually follow a chronic course associated with significantly worse prognosis and higher mortality.6,99–101 Previous findings suggest that between 20% and 90% of patients with SLE will develop some form of respiratory involvement throughout the course of their disease.101–103
As in other rheumatic diseases, prevalence rates for lung involvement in SLE vary according to study population and detection methods.3,6,102 Pleural involvement is the most common pulmonary manifestation. Interstitial lung disease is rare, particularly as an initial manifestation, and it is usually less severe than in other CTDs.2,6,101 About 1% to 15% of SLE patients have diffuse ILD or chronic pneumonitis, with NSIP being the most frequent pattern.2,3,6,8,99,100,104–107
The prevalence of SLE-ILD is higher in men and increases with age and disease stage.2,56,106,108,109 Additional risk factors associated with the development of ILD include previous episodes of acute lupus pneumonitis, Raynaud phenomenon, gastroesophageal reflux disease, tachypnea, abnormal nailfold capillaries, elevated CRP, and anti-Sm and anti-U1-RNP seropositivity.2,59,106,107 ILD is a predictor of poor prognosis.2,56,109–111
Systemic lupus erythematosus-associated ILD screening and monitoringRoutine clinical assessment of patients with SLE should include careful evaluation of respiratory involvement. Symptoms such as dyspnea, chest pain, reduced exercise tolerance, cough, and hemoptysis should prompt a search for an underlying lung disease.101,112
Diagnosis of SLE-ILD can be made by HRCT, which confirms the presence of ILD and helps to classify disease patterns and extent.59,101,102,113 Pulmonary function tests can be used to document the extent and progression of ILD.59,101,107 Autoantibodies should also be investigated, since patients with anti-La/SSB, anti-Scl-70, and anti-U1RNP antibodies are more likely to develop ILD.59,101,113
For patients considered at higher risk for developing SLE-ILD, intermittent screening with PFTs including spirometry and DLCO is recommended.114
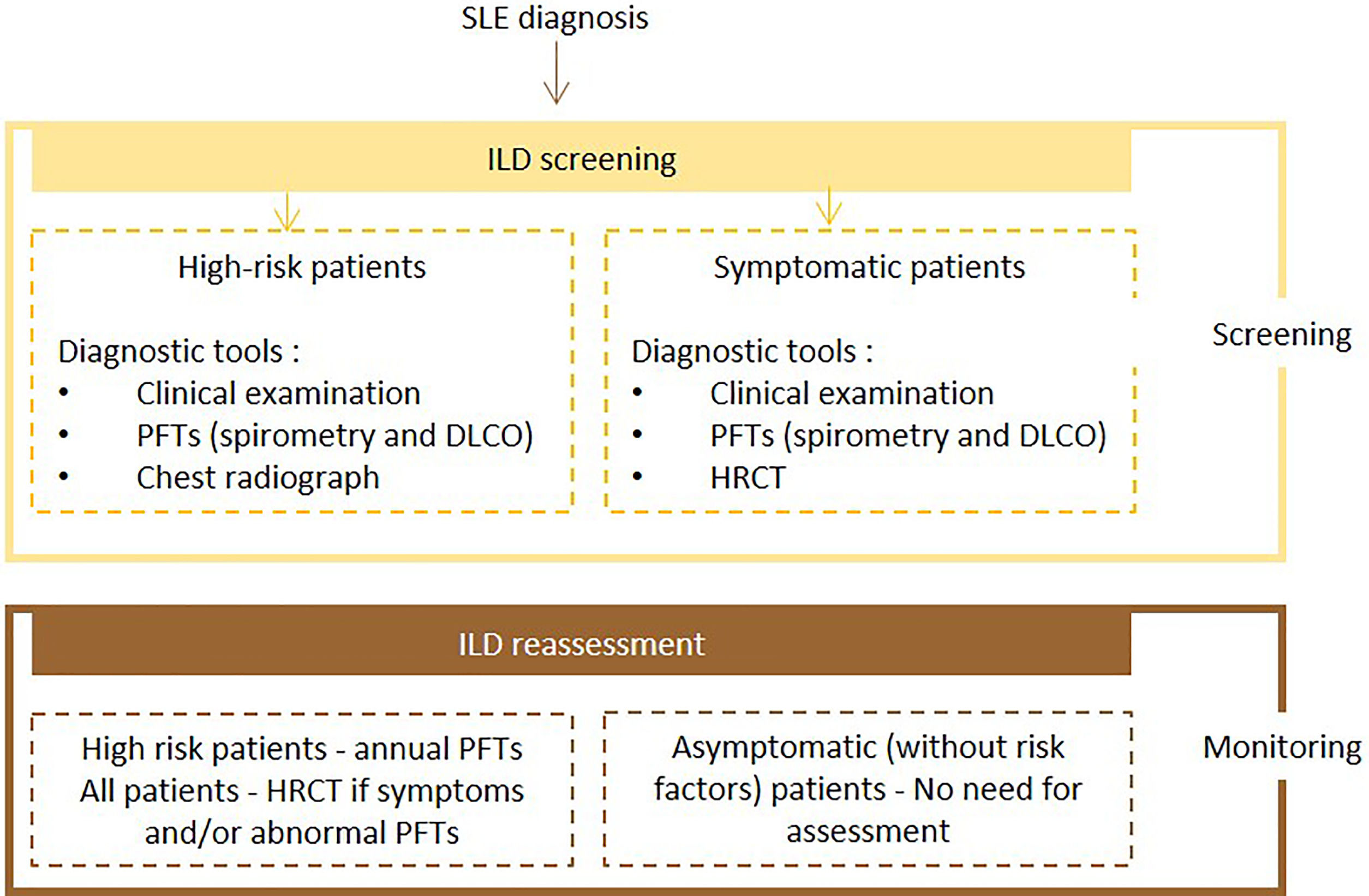
Expert panel recommendationsSymptomatic patients with SLE should undergo screening with PFTs (spirometry and DLCO) and HRCT. High-risk patients should undergo PFTs and chest radiography at baseline and PFTs annually. If symptoms and/or PFTs abnormalities occur, HRCT should be performed. There is no need for assessment in other cases.
Fig. 6 presents the recommended diagnosis algorithm for ILD in patients with SLE.

Proposed algorithm for ILD diagnosis at baseline and during follow-up with respective tools and repeat screening strategy for patients with SLE. SLE – systemic lupus erythematosus DLCO - diffusing capacity for carbon monoxide; HRCT – high resolution computed tomography; ILD - interstitial lung disease; PFTs – pulmonary function tests.
Since ILD is a major cause of morbidity and mortality in CTD, early diagnosis and severity assessment are crucial. Several innovative treatments with a positive impact on disease course and outcomes in CTD-ILD have been described and should be initiated following careful assessment. Guidelines are needed to assist clinicians in the early, accurate detection of ILD in this setting. Although there are international statements on SSc-ILD screening and some data on RA-ILD, there is a scarcity of information and guidance for other CTDs such as SS, MCTD, myositis, and SLE.
Drawing from evidence of the negative effects of ILD on CTD and the expert opinions of a panel of pulmonologists, rheumatologists, and thoracic radiologists, we have presented a series of recommendations for ILD screening in diverse systemic rheumatic disorders with a significant incidence of ILD. The primary goal of these recommendations is to highlight the importance of early, accurate recognition and to guide treatment decisions with the ultimate aim of improving disease outcomes and survival.
Medical writing assistance, supported financially by Boehringer Ingelheim Portugal, was provided by Prime Focus during the preparation of this article.


